


Introduction
RNA interference (RNAi) was initially proven to be a powerful method of knocking down gene activity by the introduction of long double-stranded RNA (dsRNA) molecules in invertebrates such as Drosophila and C. elegans. However, in most mammalian cells the introduction of long dsRNAs initiates a cellular interferon response that ultimately causes cell shutdown and leads to apoptosis. Different strategies have been adopted to get around this problem, all of which rely on the use of short dsRNA molecules designed to mimic the 21-23 bp short interfering RNAs (siRNAs) known to act as natural mediators of the RNAi response.
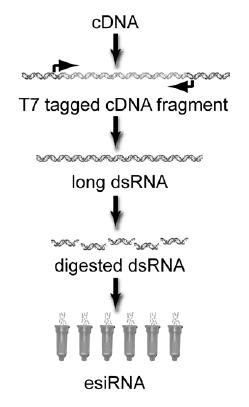
Fig 1: Flow-chart of esiRNA synthesis. Important elements and the binding sites for the T7 primers are depicted.
The most successful, best characterized and most widely used solution to date has been that of chemically synthesized siRNAs. When correctly designed, such siRNAs show good RNAi potency and specificity and, in most cases, do not detectibly activate the interferon pathway. The widespread commercial availability of high throughput RNA oligonucleotide synthesis services has assured broad accessibility of synthetic siRNAs yielding high transfection efficiencies and generally low cytotoxicity. Based on this success, several commercial siRNA libraries of siRNAs have been constructed to enable genome-wide screening in human cells. However, the inherently high cost of these reagents, which is further increased by the need to insure RNAi potency by designing multiple siRNAs for each target, currently limits their accessibility for large scale screens, especially for academic researchers.
Endoribonuclease prepared siRNA (esiRNA, Fig. 1) is an efficient, specific and cost-effective alternative to chemically synthesized siRNA that can be used for large scale screening (1). Therefore, esiRNA libraries offer a promising new approach for making genome-scale RNAi screening broadly accessible to the research community. The aim of our subproject is to carry out detailed testing and development of esiRNA libraries to complete their full maturation into a genome-scale RNAi screening technology by establishing high throughput production protocols for esiRNA. Furthermore, within the subproject a whole genome human esiRNA library will be generated to evaluate the feasibility of esiRNA library production in high throughput. Finally, esiRNA resources will be made available through the RZPD.
Project Status
For our initial screen using esiRNA (1), the generation of the esiRNA library was relatively cumbersome and time consuming. However, we have realized that many of the steps for the production can be streamlined and automated. We have tested several methods to adapt the esiRNA production to 96-well format. Several 96-well matrix plates were compared and diverse centrifugation protocols were tested. We have now established a protocol allowing the production of 2400 esiRNAs/day starting from T7-tagged PCR fragments. This enables us to produce a genome-wide esiRNA library (~ 20.000 genes) within about two weeks. We have also included several steps of quality control into the production cycle. Quality control includes three gel checks (see also Fig. 3) after the PCR reactions, after the RNaseIII digestion, and after the normalisation of the library. The normalisation is performed fully automated with a Tecan robotic workstation. The set-up and programming to run the normalisation was developed as part of this project. To allow tracking and database storage of the acquired data we have developed a LIMS system. The costs for the first generation esiRNA library were already considerably cheaper than a library of chemically synthesised siRNAs. We have further reduced the costs to about 3,- Euro/esiRNA by developing an in vitro transcription protocol with in house produced T7-polymerase. With this protocol we achieve similar dsRNA yields when compared to commercially available in vitro transcription kits. Taken together, the steps put in place affordable, high throughput, high quality and systematic tracking of esiRNA libraries.
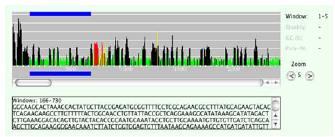
Fig 2: The webserver DEQOR allows the identification of efficient silencers and cross-silencers using state-of the-art siRNA design criteria. The in silico analysis of a given mRNA sequence is graphically displayed: efficient silencer windows (green), inefficient silencer windows (black), perfect cross-silencer windows (red), and imperfect cross-silencer windows (yellow). Suitable sequence regions can be dragged (depicted by the blue bar) and copied from a field displaying the marked sequence for subsequent primer design.
The initial esiRNA screen demonstrated the efficiency and specificity of our approach (1). However, a detailed comparison of chemically synthesised siRNAs and esiRNAs is important to document the advantages and disadvantages of each of these silencing triggers. An important difference between siRNA and esiRNA is that with the former silencing trigger all dsRNA molecules are composed of the same sequence. In contrast, esiRNA is a complex mixture of different dsRNA molecules. The complex mixture of dsRNA molecules closely reflects the natural mechanism of RNAi (e.g. viral defense) and also mimics the RNAi resources used in plants, Drosophila and C. elegans, where typically long dsRNAs are used as silencing triggers.
To investigate the knockdown efficiency of chemically synthesised siRNA and esiRNA we have chosen 50 transcripts and have designed two independent siRNAs and two independent esiRNAs targeting the 50 transcripts. The esiRNAs have been made and the siRNAs have been ordered. These silencing triggers will be transfected into HeLa cells and the knockdown efficiency will be evaluated by QPCR. Recently, the reliability of RNAi in mammalian cells has been challenged. Several groups have reported off-target effects when siRNAs were transfected into mammalian cells. Typically, cross-silenced transcripts showed some degree of homology to the siRNA sequence, indicating that a perfect match of the siRNA sequence to the targeted transcript is not required. To investigate the differences in off-target effects between silencing with siRNAs or esiRNAs we have designed three different siRNAs and three different esiRNAs against three different transcripts. Gene expression profiling experiments will be performed to compare the different silencing triggers. These experiments will be performed with esiRNA generated with E. coli RNaseIII and purified Dicer.
Generation of a second generation genome-wide esiRNA library
Our initial esiRNA library was generated from IMAGE-clones that carried a gene specific cDNA insert. While this source as the PCR template is very convenient because universal primers can be used to amplify the insert, it also has some disadvantages. The main disadvantage is the fact that the sequence used for the generation of the esiRNA is fixed. However, transcripts may contain regions which contain a higher proportion of potentially good silencing triggers than other regions. To map good silencers onto transcripts we have previously developed the program DEQOR (2). This program (http://deqor.mpi-cbg.de/) produces a „heat-map” of predicted good versus bad siRNAs (see Fig. 2). A region of the transcript that contains a large number of good silencers has a higher chance of generating a more effective esiRNA.
We have performed a genome-wide DEQOR analysis of all human protein coding transcripts and have developed an algorithm that automatically identifies an optimal region for esiRNA production. Sequence specific primers to all these regions were designed. An important question was which template source could be used to amplify the fragments? For similar Drosophila and C.elegans libraries genomic DNA was used. Because human genes typically contain large introns and short exons this option was not suitable. We tested a cDNA source and obtained the predicted bands for a large number of transcripts (see Fig. 3). Therefore this method is suitable for the generation of a genome wide esiRNA library. 40.000 gene specific oligos have been ordered and the production of the genome-wide esiRNA library is underway.
Distribution of esiRNA resources
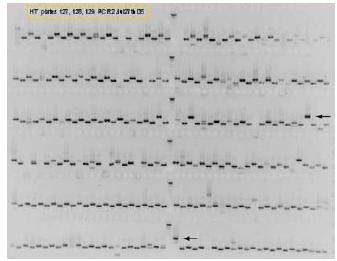
Fig 3: A typical agarose gel showing gene specific PCR fragments generated using a cDNA resource as template source. Loading was performed such that an alternating band size pattern indicates the correct amplicon. Note that few bands show a band size that is different from the prediction (arrows). These may indicate wrong annotation of these genes in the public databases or PCR artifacts.
The esiRNA resources developed in subproject 1 will provide scientists with a cost-efficient alternative to chemically synthesized siRNAs. Products will be integrated into RZPD’s search tools and distribution infrastructure. Researchers will have access using gene-based information, sequence information or download the complete dataset established in this subproject from the respective webpage (www.rzpd.de/products/esiRNA/).
Outlook
RNAi methodology in general and large scale RNAi screens in mammalian cells, in particular is an important technology for the functional dissection of the mammalian genome. RNAi experiments offer relatively inexpensive loss-of-function analyses which will help to decipher the function of genes, including the genes implicated in human disease. However, it is important to identify the appropriate silencing triggers for an experiment. This subproject will give insights into the different silencing triggers that can be utilised in RNAi experiments and will deliver a cost effective alternative to chemically synthesised siRNAs. This resource also aims to enable scientists to perform affordable genome-wide RNAi screens in mammalian cells
Lit.1: Kittler R et al. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature. 2004 May 23;432(7020):1036-40. 2. Henschel A et al. DEQOR: a web-based tool for the design and quality control of siRNAs. Nucleic Acids Res. 2004 Jul 1;32:W113-20