

Introduction
The modulation of protein-protein interactions by small organic molecules represents one of the most rewarding yet challenging topics of current research at the interface of organic chemistry and biochemistry [1]. Since the biological function of most proteins depends on their interactions with other macromolecules, disruption or enhancement of these interactions by cell-permeable molecules provides a means to influence protein function. Cell-permeable molecules which allow a given protein to be turned on or off with high temporal control may serve as lead structures for drug development. However, the following difficulties need to be overcome: 1) protein-protein interfaces are significantly larger than the surface areas of small molecules, 2) many protein-protein interfaces lack obvious binding pockets for small molecules, and 3) mechanism-based or natural product-based lead structures rarely exist.
We aim to target the protein-protein interactions required for signaling through the transcription factors Stat3 and Stat5, as well as the kinase Plk1, all of which are regarded as candidate targets for therapeutic treatment of certain human cancers. The conventional approach to inhibit these essential mediators of signaling involves the use of small-molecule inhibitors of kinase activity specific to either upstream kinase activity which phosphorylates Stat3/Stat5, or to the kinase activity of Plk1 itself. This approach suffers from the following two drawbacks: firstly, current small molecule kinase inhibitors target the ATP-binding site which is conserved throughout the kinase families, therefore it is not surprising that these inhibitors usually act on more than one single kinase. Secondly, as the kinases which activate Stats are involved in other signaling pathways as well, their inhibition invariably leads to undesired side effects. In contrast, small molecules inhibitors of the less conserved protein-protein interactions required for Stat activation and/or homodimerization, or for the binding of Plk1 to its substrates, have the potential to exert more specific effects than kinase inhibitors. Therefore, their use as therapeutics is likely to be accompanied by fewer side effects.
Results/Project Status
Stat3 is constitutively active in a wide range of primary human tumors (eg. various leukemias; breast, head and neck, brain and prostate cancers; multiple myelomas; lymphomas). It is strongly expressed in certain colon cancer-derived cell lines. Stat3 appears to act as an essential mediator of dysregulated upstream tyrosine kinase activities frequently observed in cancer cells. Inhibition of constitutively active Stat3 by a dominant negative Stat3-mutant or using antisense approaches results in growth inhibition and apoptosis of tumor cell lines. A peptide which binds to the SH2 domain of Stat3 inhibits Stat3 homodimerization in vitro has been shown to block v-src-induced gene transcription and cell transformation in tissue culture.
Stat5 is overactive in several kinds of leukemias (eg. HTLV-dependent, erythroleukemia, acute lymphocytic leukemia, acute myelogenous leukemia, chronic myelogenous leukemia). The oncogenic potential of Stat5 was demonstrated by the identification of a Stat5-mutant with transforming properties. Constitutively active Stat5 is essential for transformation by Bcr-Abl, and inhibition of Stat5 by a dominant negative mutant or by an inhibitor of Bcr-Abl kinase activity abrogates cellular transformation by Bcr-Abl.
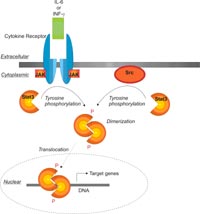
The serine/threonine kinase Plk1 (polo-like kinase 1) is overexpressed in various human tumors (eg. breast or colorectal cancer) and has been implicated as an adverse prognostic marker for cancer patients [2]. Its inhibition in cancer cells by small interfering RNA or by a Plk1-derived peptide (Antennapedia-polo-box-fusion) that inhibits phosphorylation of Plk1 substrates was demonstrated to inhibit proliferation of various tumor cell lines by inducing mitotic arrest and apoptosis. Antisense oligonucleotides targeted against Plk1 were furthermore able to inhibit tumor growth in vivo. As the interaction between Plk1 and its substrates is mediated by the carboxyterminal portion of Plk1 containing the polo-box domain (PBD) of Plk1 (which is only present in Plks) and a novel S-(pT/pS)-(P/X) tripeptide motif in its substrates, inhibitors of binding can be expected to be more specific than ATP-binding site inhibitors. These compounds would present the first small molecule inhibitors of a serine/threonine kinase which act by inhibiting the protein-protein interaction between the kinase and its substrates.

Inhibitors of Stat3
Activity of STAT3 requires its own SH2-domain-mediated binding to phosphotyrosine-containing sequences. We have developed a high-throughput binding assay, based on fluorescence polarization, which allows screening for small molecules that bind to the STAT3 SH2 domain and thereby inhibit its activity [3]. The basis of this assay is the binding of a fluorescein-labeled phosphotyrosine–peptide derived from the interleukin-6 receptor subunit gp130 to unphosphorylated STAT3 with a Kd of 150 nM. The assay is stable with regard to salt concentration, dimethyl sulfoxide concentration, and time. It has been adapted to a 384-well format, with a Z’ value of 0.87, and can be used to screen for small molecules that bind to the STAT3 SH2 domain. By screening structurally diverse chemical libraries consisting of a total of 17,298 cell-permeable small molecules, we identified 9 compounds which inhibited the interaction between unphosphorylated Stat3 and a fluorescein-labeled phosphotyrosine-containing peptide by more than 90% (at 100 µM compound concentration). 3 of these compounds inhibit dimerization and subsequent DNA-binding of purified, phosphorylated Stat3 by 59-95% at 50 µM compound concentration. We are currently in the process of analyzing the specificity of the Stat3 dimerization inhibitors we have identified. To this end, we will investigate their effect on other SH2-domain mediated protein-protein interactions. We have already established that the chemicals do not generally inhibit protein-protein interactions.
We are currently in the process of analyzing both potency and specificity of the 3 compounds’ abilities to inhibit nuclear translocation by immunofluorescence using antibodies against Stat3 and other Stats. Nuclear translocation of Stats in the presence of the inhibitors will also be analyzed in collaboration with Dr. R. Stauber, who has developed a system which allows the analysis of nucleo-cytoplasmic transport by microinjection of a GFP-tagged protein [4]. The effects of the compounds on Stat3 activation are currently monitored in proliferation assays, reporter gene assays and in focus formation assays. In order to analyze the global specificity of the compounds, we will synthesize derivatives which can be immobilized on a resin whilst retaining their activity. After incubation of the immobilized inhibitors with cell lysates, binding proteins will be identified by mass spectrometry. In collaboration with Professor J. Sleeman we plan to test the compounds for their effect on tumor growth in mouse models for colorectal and breast tumors which display constitutive activity of STAT3. The collaboration with Professor G. Hämmerling is aimed at analyzing the inhibitors’ effects in inducible autochthonous tumor models which co-express luciferase and thereby allow in vivo imaging of tumor growth.
We will utilize the Stat3 dimerization inhibitors as tools to study the effect of downregulation of constitutively active Stat3 on tumor gene expression profiles. Although reversion of tumor phenotype has been shown to result from Stat3 inactivation by dominant negative mutants and antisense studies in MO7e cells (acute myeloid leukemia) and DU-145 cells (prostate carcinoma), as well as the use of the JAK2 inhibitor AG490 in Hodgkin’s tumor cell lines, the effect of inactivating Stat3 on gene expression profiles in cancer cell lines has not been investigated. The reduction of Stat3 activity in different cancer cell lines using dimerization inhibitors can be used to identify Stat3 target genes in each of these cell lines. In addition, the extent to which gene expression in these tumor cells after inactivation of Stat3 resembles the profile of gene expression by cell lines derived from corresponding normal tissue can be assessed. We plan to analyze gene expression in various cancer cell lines which contain constitutively active Stat3 in the presence or absence of the Stat3 inhibitors, and compare the profiles of gene activation with those of cell lines derived from normal tissue. The Stat3 inhibitors will also be analyzed in collaboration with Prof. A. Ullrich for their effects on chemoresistant tumor cell lines with constitutively active Stat3.
Inhibitors of Stat5
We aim to establish a high-throughput binding assay-analogous to the assay developed for Stat3- to monitor binding of compounds to the SH2 domain of Stat5, and screen chemical libraries for Stat5 inhibitors. Screening hits will be validated and characterized in independent in vitro assays and cellular assays. Further analysis will be performed in collaboration with Professors B. Groner and L. Hennighausen (NIH, Maryland, USA), Dr. R. Stauber (analysis of nucleo-cytoplasmic transport), as well as Professors G. Hämmerling and J. Sleeman (mouse tumor models).
Inhibitors of Plk1
We are working on an assay amenable to a high-throughput format which will allow us to analyze the ability of small molecules to disrupt the interaction between Plk1 and the optimal recognition sequence of its substrates. Screening hits will be analyzed for their ability to inhibit the interaction between Plk1 and some of its substrates (eg. Cdc25c, Cyclin B1) in ELISA. In vitro kinase assays will be used to confirm that the inhibition of enzyme-substrate binding correlates with reduced Plk1 serine/threonine kinase activity, and will be employed to analyze the inhibitors’ specificity for Plk1. In collaboration with Professor K. Strebhardt, the most potent and specific Plk1 inhibitors will be analyzed for their effect on cell cycle distribution, apoptosis, and cancer cell proliferation in cellular assays and in vivo. Further analysis of the inhibitors will be performed in Professor G. Hämmerling’s mouse tumor models.
Outlook
The successful completion of the projects will lead to the identification of cell-permeable, selective, small-molecule inhibitors of 3 established cancer targets: Stat3, Stat5 and Plk1. These substances will be useful tools for improving our understanding of the role of the respective proteins in oncogenesis and may provide lead structures for drug development. Furthermore, the analysis of structure-activity relationships in combination with structural investigations will improve our understanding of how small molecules can interfere with the association of large protein-protein interfaces. The cell based translocation screening assays we developed in collaboration with the SMP Cell, will catalyze the accomplishment of our research tasks and are available to all NGFN members.
Lit.: 1. Berg T. Modulation of protein-protein interactions with small organic molecules. Angew Chem Int Ed Engl. 2003 Jun 6;42(22):2462-81. 2. Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005 Jan 10;24(2):267-76. 3. Schust J, Berg T. A high-throughput fluorescence polarization assay for signal transducer and activator of transcription 3. Anal Biochem. 2004 Jul 1;330(1):114-8. 4. Knauer SK, Moodt S, Berg T, Liebel U, Pepperkok R, Stauber RH. Translocation biosensors to study signal-specific nucleo-cytoplasmic transport, protease activity and protein-protein interactions. Traffic. 2005 Jul;6(7):594-606.