

Introduction
Gliomas are the most frequent primary tumors of the brain. They are classified into several histological entities and malignancy grades. Molecular analyses have shown that specific genetic lesions are associated with the different types and malignancy grades of gliomas. Furthermore, certain genetic alterations, in particular allelic losses on chromosome arms 1p and 19q, have been shown to represent powerful predictors of prognosis and response to therapy in oligodendrogliomas. However, the relevant target genes on these and several other chromosomes frequently affected in gliomas are still unknown. Differential methylation of cancer associated genes plays a pivotal role in tumor development. Several studies, either using candidate gene analyses or genome wide approaches, suggest that these alterations may occur at different stages during carcinogenesis. A study using restriction landmark genomic scanning, which allows for the global analysis of the genome methylation status, has estimated that an average of 600 de novo methylation events occur within a single tumor genome. Moreover, hypermethylation of CpG islands develop in a non-random fashion and show high specificity for different tumor types (1). Candidate gene approaches have shown that hypermethylation of CpG islands in gliomas may cause functional inactivation of different tumor suppressor genes. These include TP73, MGMT, CDKN2A, CDKN2B, p14ARF, Peg3, RB1, hMLH1, THBS1, N33, HIC1, c-abl, and c-fos. Recently it has been shown that hypermethylation of the MGMT CpG island is a useful predictor of responsiveness of glioblastomas to alkylating agents supporting the relevance of epigenetics in gliomas (2). To investigate the role of epigenetic DNA alterations in glioma pathogenesis at a comprehensive level, we employ a microarray-based technology known as differential methylation hybridization (DMH) to determine the methylation status at 7776 CpG islands.
Results
Epigenetic profiling of glioma entities
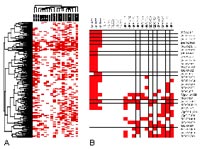
In our initial studies DMH was used to globally profile CpG island hypermethylation in 20 gliomas and 4 normal (white matter) controls (3). These gliomas were categorized into three subtypes defined by the World Health Organization (WHO) according to tumor behavior and histology: pilocytic astrocytoma PA WHO grade I, oligoastrocytoma OA WHO grade II, and glioblastoma GBM WHO grade IV. Of the 7,776 CpG islands screened, 763 loci were identified to be hypermethylated in at least one glioma sample relative to the control. The overall methylation frequency of the individual tumors ranged from 0.3 to 2.4% (18 to 160 hypermethylated loci). As shown in Fig 1, the overall methylation event is significantly less in the low-grade PAs as compared to OAs and GBMs (0.45% verses 2.0% and 1.4%).
Hierarchical clustering of the 763 methylated loci clearly separated the 20 gliomas based on their clinicopathological parameters and WHO grading. A subpanel of 20 loci hypermethylated in advanced gliomas (OAs and GBMs) was selected for sequence analysis (Fig 1b). Within the subpanel, 8 loci were preferentially methylated in GBMs, six of which contained sequences identical to known genes (AGP2, Cllorf23, SMARCA5, HoxA10, EML-2, and FoxD3). To substantiate these microarray findings, methylation-specific PCR was conducted in an expanded set of gliomas (PA, AIII, O, OA, and GBM). The identified CpG island spanning the promoter and first exon of SMARCA5, a known chromatin modulator, showed de novo methylation in 91% (10/11) of grade IV and 20% (5/26) of grade III tumors, respectively. No ypermethylation of this locus was seen in low-grade (grade I or II) gliomas (0/21). Thus the identified sequence can be used as a sensitive epigenetic marker of malignancy in gliomas. In addition we have described 10 CpG island loci uniquely hypermethylated in the OA group. Among these, the sequences of 4 loci matched to known genes or cDNAs, including PRKD2, MSF, MGC5627, and C31PI. Another sequence that was found to be exclusively methylated in gliomas with oligodendroglial differentiation was identical with a CpG island located on chromosome14q32. In line with the microarray data, MS-PCR revealed hypermethylation of this locus in 80% (8/10) of OA, but in none of the PA (0/8) or GBM (0/8) samples analyzed. In addition, we found that the majority of oligoastrocytomas and 40% of oligodendrogliomas displayed either allelic loss in these regions or hypermethylation of the 14q32.12 locus. It is therefore conceivable that this chromosomal region may contain a critical tumor suppressor, the expression of which can be inactivated via genetic and/or epigenetic alteration in gliomas with oligodendroglial differentiation. As such, the epigenetic markers uncovered by this approach may complement current histopathological classification for disease diagnosis and grading.
Epigenetic silencing of PCDH#A11 in diffuse astrocytomas
To identify targets for de novo methylation involved in the initial steps of transformation of astrocytic gliomas we have generated an epigenetic profile of astrocytomas (WHO grade II). Among the genes identified so far, we have described extensive de novo methylation within a CpG island spanning promoter and first exon of the PCDH-#-A11 gene, a cell-cell contact molecule of the nervous system (4). Based on the function of the gene product, it is tempting to assume that transcriptional inactivation of this gene may affect the adhesion of glial cells to neighboring cells thus promoting invasive growth, a hallmark of astrocytomas and a major obstacle to neurosurgical intervention.
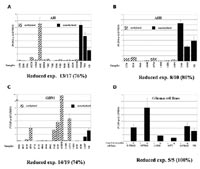
We conducted a detailed methylation analysis of this region in 57 primary astrocytic tumors and eight glioma cell lines by bisulfite sequencing (Fig 2a) and COBRA analysis. Hypermethylation was detected in 88% of astrocytomas (WHO-grade II and III), in 87% of glioblastomas WHO-grade IV and in all analyzed cell lines. There was a highly significant correlation (p=0.00028) between PCDH-###11 hypermethylation and decreased transcription as determined by competitive RT-PCR in WHO-grade II and III astrocytomas. After treatment of glioma cell lines with a demethylating agent, transcription of PCDH-###11 was restored confirming transcriptional silencing by DNA methylation (Fig 2b).

Outlook
The current classification of central nervous system tumors is based on the origin of the putative tumor cells and histopathological features according to the WHO grading system. Classification and grading, however, can be subjective and there are considerable variations in treatment outcome in patients with similar histopathological features. Moreover, biological players responsible for tumor biological hallmarks of most gliomas entities like invasion of normal brain tissue and resistance to chemotherapy, the major obstacles for a successful treatment, are still unknown. Our present data provide proof of principle that CpG island hypermethylation is a potential source of biomarkers for different gliomas entities. Within this project we are going to establish a comprehensive epigenetic profile of all major glioma entities. Our large-scale approach of DNA methylation analysis will reveal novel genes affected by aberrant methylation in gliomas. Finally, the identification of distinct methylation signatures will provide novel insights into the molecular basis of tumor formation, which will be useful for tumor classification, as well as prediction of response to therapy and prognosis. The principle reversibility of epigenetic silencing opens up new possibilities for the treatment of glioma patients.
Lit.: 1. Costello JF et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000 Feb;24(2):132-8. 2. Hegi ME et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005 Mar 10;352(10):997-1003. 3. Waha A et al. Methylation profiling identifies epigenetic markers for high-grade gliomas. Cancer Genomics and Proteomics. 2004 (1), 209-214. 4. Waha A et al. Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia. 2005 (7), 193-199.