

Introduction
Novel molecular data, such those from gene expression or protein profiling are high-dimensional and require special methods both with regard to storage and to bio-statistical analysis. Our database application for the maintenance of microarray data, iCHIP (http://www.dkfz.de/ibios/ ngfnServices), is installed at several locations within the clinical networks. In addition, it has also been established as a local decentralized version. The conceptual design of iCHIP is in accordance with the MIAME- and MAGE-standards (1) of the international MGED consortium. Originally iCHIP was developed to function as a gene expression database suitable for data generated from DNA array screening, using both oligonucleotides and cDNAs. iCHIP has been flexibly and modularly constructed, making iCHIP very much an analysis toolbox.
With regard to analysis, we have developed workflows to deal with the high dimensionality and low sample numbers. Such workflows, based on nested cross-validation, have been implemented in a Bioconductor package based on the statistical environment R (www.r-project.org). A typical application is classification analysis, where patterns are determined that allow for accurate prediction of a given class distinction. That is, given a set of training data of known classes, algorithms like support vector machines are trained to predict these classes in new data sets. The methods may be combined with feature selection procedures like recursive feature elimination to obtain comprehensive patterns with high discriminative power.
Typical questions to address in the context of glioblastoma would be, which genes change expression in samples with high expression compared to those with low expression, or which genes are associated with good or bad prognosis, or which genes are associated with established markers like MYC amplification, advanced age or deletion of specific chromosomal region.
Results/Project Status
Extension of iCHIP database
The iCHIP database was originially designed to store data from gene expression profiling studies based on cDNA microarray or oligonucleotide array technology. We have extended the database to be able to deal with tissue microarray, single nucleotide polymorphism and proteome profiling data based on SELDI technology. The database is already partially filled with such data, including the most relevant associated clinical data of those patients whose tumor specimens have been investigated. Access restrictions and security guidelines prevent unauthorized access to the data, which are stored in anonymous form. A web-based front-end to the database exists to facilitate interaction with the database (Fig. 1). Export to flat files and reports are available. Based on the general iCHIP database and application, a specific glioblastoma database was established. As a starting point, 18 expression profiling data (see expression analysis below) from primary glioma tumors were collected within this database.
Analysis of gene expression data
Modern tumor research requires fast and specific diagnosis tools. For tumor patients it is of primary interest to know which state their disease has reached and what therapies may be best suited. Therefore, understanding the cellular processes is an essential precondition.
Over the past few years, scientists have established many high throughput methods in Molecular Biology from different biological levels (genome, transcriptome, proteome). This provides us with an opportunity to understand cellular processes at a level heretofore impossible. In one of our main projects we will develop methods for using information on genomic aberrations (array-based CGH) (2), DNA methylation (3), gene expression levels (DNA- Micro-arrays) (4) and proteome composition (2D gel electro-phoresis) (5) to find markers that are indicative of certain disease states. We use different data types simultaneously and thereby focus on inherent dependency structures that are given by the processes of transcription and translation. We expect that by using all information simultaneously, we will significantly improve molecular diagnostics in applications where use of one method alone does not yield sufficient accuracy.
In this project we are analysing chromosomal losses and gains (CGH-Data) and their correlation with over/underexpression of genes (DNA-Microrarray-Data) belonging to specific chromosomes. The integrative analysis of genomic and transcriptomic data was applied to examining cases of spontaneous Rezidivation in primary Glioma. The data were derived from an identical series of 9 patients with low level malignant Glioma (WHO-Rating II) which spontaneously form malignant relapses (WHO-Rating III or IV). Altogether we had 18 tumors from 9 Patients that were analysed by CGH and DNA Microarray Technology.
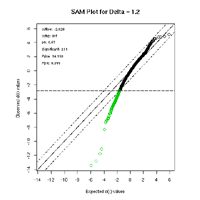
At present, we could show that the group of relapsed tumors had 231 differently expressed genes, see figure 2. In further investigations we have to clarify the coherence of our present results coming from the transcriptomic level and the results of analysing the genomic level, which is still in process.
Outlook
We will extend the work on the iCHIP database to include more experimental data, better annotation of novel data types (SNP, proteomics data, tissue micorarray data), extended front-ends to the database, export of MAGE-ML data, and connection to the integrative bioinformatics system BioRS (BioMax, Munich). Besides this technological iCHIP extension, the collection of high-throughput data from more glioma tumors will be completed, in order to enable integrative cross-platform analyses. With respect to such analyses, we will continue our bio-statistical analyses on new data arising from other work packages. We will use methods of meta-analysis and cross-platform analysis to obtain a consistent view on all publicly available data together with those of this project. The presently analyzed glioblastoma collection will be upgraded to around 100 tumor samples from same study. More data available from other collaboration partners within the CCN network will round off the glioma collection. This will lead to an improved understanding of glioma progression, spontaneous regression, and resistance to therapy. Proteomics data will help to contrast existing gene expression profiles and to understand changes at the protein level.
Lit.: 1. Ball, C.A., Sherlock, G., Parkinson. H et al.., Microarray Gene Expression Data (MGED) Society. Standards for microarray data. Science.. 298:539 (2002). 2. Lichter, P., Joos, S., Lampel, S. (2000) Comparative genomic hybridization. Uses and limitations. Sem. Hemat. 37, 348-357 3. C. Mund, V. Beier, P. Bewerunge, M. Dahms, F. Lyko and J.D. Hoheisel (2005) Array-based analysis of genomic DNA methylation patterns of the tumour suppressor gene p16INK4A promoter in colon carcinoma cell lines. Nucleic Acids Research, Vol.33, No.8 4. DJ Lockhart and EA Winzeler (2000) Genomics, gene expression and DNA arrays. Nature, 405(6788):827-836. 5. Patel K, Stein R, Benvenuti S, Zvelebil MJ (2002) Combinatorial use of mRNA and two-dimensional electrophoresis expression data to choose relevant features for mass spectrometric identification. Proteomics Oct;2(10):1464-73.