

Introduction
Neuronal cells (neurons) are widely branched and connected in a huge network within the brain as well as to many other cells in the body, such as muscle cells. Neurons conduct signals along their extensions via so-called action potentials. To communicate with connected neurons or different cell types, they form synapses, in which a chemical transmitter is released presynaptically and activates a postsynaptic receptor. Such a synaptic transmission can be either excitatory or inhibitory. The basis of these fundamental skills of neurons is an electrical excitability which is provided by proteins called ion channels. Ion channels form pores within the neuronal cell membrane that selectively conduct specific ions such as Na+, Cl-, K+ and Ca2+. They react to specific signals like membrane voltage changes or transmitter release with opening and closing. Voltage-gated Na+ and K+ channels are responsible for the generation and propagation of action potentials, whereas synaptic signal transduction is mediated via ligand-gated ion channels such as nicotinic acetylcholine receptors or gamma-amino-butyric-acid (GABA) receptors. Various other channels, like Ca2+ and Cl- channels, pre- or postsynaptically regulate intra- and extracellular ion concentrations and their transmembrane gradients, which as well are key players of excitatory and inhibitory signals in the nervous system.
Epilepsy is defined as a disease of the brain with recurring epileptic seizures. Seizures occur, when signals in the nervous system get unbalanced. Then a group of neurons becomes hyperexcitable and starts to fire in a synchronized way. This abnormal activity can spread to other brain regions and become generalized. Depending on the brain regions involved in seizure activity, patients will have different symptoms such as shaking of an arm, bahavioral arrest or severe generalized convulsions. Due to the basic principles of excitability and ion channel function, the symptomatic drug therapy of epilepsies is mediated via the modulation of ion channels that can increase inhibition or block excitation of neurons.
The underlying causes of epilepsy can be symptomatic as with brain tumors, strokes or scars, or genetic or both. Genetic factors are expected in at least 40% of all epilepsies. We are interested in pure, inherited forms of epilepsy (so-called idiopathic epilepsies) that are not associated with brain lesions. The general aim is to identify the genetic causes and characterize their functional consequences. This should enhance our understanding of the genesis of epilepsy and on a longterm lead to better therapies.
Following the aforementioned principles of ion channel function and the good response of the epilepsies to treatment with ion channel modulators, it appears as a logical consequence that an increasing number of mutations has been detected in ion channel encoding genes within the recent 10 years, which cause different forms of the idiopathic epilepsies. Most of these syndromes are rare monogenic diseases, but recently, mutations have also been detected in common epilepsy subtypes. For example, a generalized epilepsy with febrile seizures (GEFS+) and the most severe form of this syndrome, severe myoclonic epilepsy of infancy (SMEI), are caused by mutations in two subunits of the voltage-gated Na+ channel, the most important channel to initiate and conduct action potentials, or in a GABAA receptor subunit), the most important inhibitory synaptic receptor in the brain. A mutation in another GABAA receptor subunit gene has been detected in a family with one of the most common epilepsies, juvenile myoclonic epilepsy (an actual overview of genetic alterations in idiopathic epilepsies is given by Lerche et al. 2005).
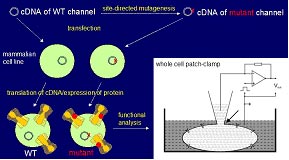
Results
Methods used
Our part within the national genome project is to identify the functional consequences of epilepsy-associated mutations in ion channels that have been identified by the genetic groups of our network. The first step in the characterization of a mutation is to introduce it in the gene that can be delivered to a cell in which the abnormal function can be studied. We use an artificial system for this purpose which does not express itself (endogenously) the same channel subtype so that we have a pure population of the channel of interest. When we deliver the non-mutated (wild type) channel in one population of these cells and the mutated channel into another one, we can study both independently and compare their properties in a very detailed way. The technique we use is the so-called patch clamp technique: a glass pipette is brought to the cell surface under microscopic control to form an electrical connection with the cell allowing the study of the gating properties of these channels, that is their openings and closings under certain conditions such as changes of membrane voltage (Fig. 1). In this artificial system we generate the first hypotheses as to how a mutation can cause epilepsy. In a second step, we are interested to deliver these mutations also to neurons and test our hypotheses under more natural conditions.
Recent Studies within the Project
In collaboration with Dr. Armin Heils, who performed the genetic studies, we found and characterized novel mutations in several different genes that were associated with the most common forms of genetic epilepsies, the so-called idiopathic generalized epilepies comprising absence epilepsies, juvenile myoclonic epilepsy and epilepsy with generalized tonic-clonic (Grand-Mal) seizures on awakening.
We identified three distinct mutations in a Cl- channel gene in three independent families representing the whole spectrum of idiopathic generalized epilepsy. This channel shuffles Cl- ions out of a neuron and therefore plays an important role in establishing and maintaining a high transmembrane Cl- gradient, with a low Cl- concentration inside and a high outside the cell. This Cl- gradient is essential for the inhibitory response of GABA receptor channels. The mutations revealed different disease-causing mechanisms. The main one leads to a loss-of-function of the channel predicting an intracellular Cl- accumulation which interferes with the inhibitory action at GABAergic synapses. When inhibition is blocked within the brain, neurons become hyperexcitable and this can perfectly explain the occurrence of epileptic seizures. Thus, we here described a novel epilepsy gene, the first one that can be associated with all common suptypes of idiopathic generalized epilepsies, and elucidated the disease-causing mechanism which is an indirectly disturbed GABAergic inhibition (Haug et al. 2003).
There is also direct evidence that GABAergic synaptic inhibition is genetically impaired in idiopathic epilepsies. As already mentioned in the introduction, mutations have been detected in GABA receptor encoding genes in generalized epilepsy with febrile seizures but also in juvenile myoclonic epilepsy (JME) (Cossette et al. 2002). We now analyzed the JME-associated mutation in molecur detail including a thorough electrophysiological analysis using whole cell and single channel patch clamping with a fast application technique mimicking synaptic transmission, as well as trafficking experiments with fluorescent fusion proteins. We found a a clear loss-of-function of GABA-induced currents caused by a combination of a 30-fold reduced GABA sensitivity, faster deactivation and a largely reduced expression in the surface membrane of the mutant receptor channel compared to the wild type. In addition we could show, that this mutation probably affects the coupling between the binding of GABA to the receptor whereas activation of the receptor with barbiturates is still functioning normally (Krampfl et al. 2005), Our results thus demonstrate, that a genetically driven direct loss of inhibition can cause epileptic seizures.
Outlook
We are currently studying further mutations in other ion channels and expressing different mutations and ion channels in neurons to answer the following questions: What is the spectrum of affected ion channel genes in idiopathic generalized epilepsies and how can we explain that the different genetic alterations cause phenotypically similar seizures? Where are the affected channels exactly located in the brain and how do they function? Is there an age-dependent regulation of channel expression that correlates to and can explain the age dependence of the investigated epileptic syndromes? Do these mutations really alter the behavior of neurons in the way we predict from our studies in artificial expression systems and can this explain the epileptic phenotype in a neuronal network?
Several other mutations that were identified by Dr. Heils or Prof. Steinlein, another investigator in our network, are currently under investigation in our laboratory. They include for example genetic alterations (i) in Ca2+ channels which are important for oscillatory networks generating the typical spike and wave activity seen in the electroencephalogram (EEG) of patients with absence seizures and other types of idiopathic generalized epilepsies; or (ii) in a class of K+ channels that generate a conductance (the ‘M-current’) active in the subthreshold range of an action potential, a very potent mechanism to regulate the excitability and firing rate of many neurons.
The genetic findings in the latter, the so-called KCNQ-type K+ channels, nicely illustrate, how our studies can contribute to develop novel antiepileptic therapies. So far, we have no drug available in the treatment of epilepsy that activates a K+ channel. Such substances do exist for several years, but they also act on K+ channels located in other regions of the body as in the heart and the blood vessels leading to severe cardiovascular side effects. In parallel with genetic investigations identifying the first mutations in KCNQ channels in epileptic patients, a KCNQ channel activator, retigabine, was discovered, which only acts on K+ channels located in the brain (reviewed by Lerche et al. 2005). We recently identified the detailed molecular mechanism how retigabine opens the channel KCNQ2 channel by binding to its activation gate (Wuttke et al. 2005) which was found to be almost identical for KCNQ3 by another group in our network (Schenzer et al. 2005). Such studies raise hopes for novel concepts in the treatment of epilepsies and also many other diseases going along with a disturbed excitability of the nervous system such as migraine, neuropathic pain and stroke.
Lit.: 1. Lerche H et al. Ion channel defects in idiopathic epilepsies. Curr Pharmaceut Design 2005;11:2737-52. 2. Haug K et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet 2003;33:527-32. 3. Cossette et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184-9. 4. Krampfl K et al. Molecular analysis of the A322D mutation in the #1-subunit of the GABAA receptor causing juvenile myoclonic epilepsy. Eur J Neurosci 2005;22:10-20. 5. Wuttke TV et al. The new anticonvulsant Retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharm 2005;67:1009-17. 6. Schenzer A et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci 2005;25:5051-60.