

Introduction
Differential vulnerability of neuronal populations to cell death is found in most neurodegenerative diseases. In Parkinson�s disease (PD), this differential vulnerability is particularly striking within the clinically most relevant population of dopaminergic (DA) midbrain neurons. Here, highly vulnerable DA subpopulations within the substantia nigra (SN) and their axonal projections to dorsal parts of the striatum are almost completely lost, while neighbouring DA subpopulations within the ventral tegmental areal (VTA), and their projections to ventral striatum survive to a large extent (Damier et al., 1999).
For rare forms of familial PD, several causal gene-mutations have been identified (e.g #-synuclein, parkin, UCHL1, DJ-1, PINK1, LRRK2) but as their respective gene products are ubiquitously expressed, they alone do not explain the selective pattern of DA cell loss. For the common sporadic form of PD, diverse candidate mechanisms are discussed, including mitochondrial dysfunction, oxidative stress, and proteasomal impairment. However, as the stereotypical pattern of differential cell-loss of DA neurons is reproduced in mechanistically distinct PD animals models, different toxic and molecular pathways might converge on a common but yet unknown cellular mechanism for differential neuronal vulnerability (Greenamyre and Hastings, 2004).
The molecular and functional mechanisms of differential vulnerability of DA midbrain neurons are still unresolved. However, differences in vulnerability of DA neurons are likely to be related to cell-specific differences in function and gene-expression; more resistant DA neurons might either constitutively express a specific subset of genes that confer resistance to PD-inducing trigger factors, or they might alter gene-expression in response to a neurodegenerative trigger.
Thus, we aim to identify genes and related mechanisms - on a basis of an identical genome - that are differentially expressed in individual DA neurons displaying differential vulnerability in response to Parkinson�s disease trigger factors. Our approach is complementary to clinico-genetic population studies that aim to identify gene variations associated with an altered PD-risk. In cooperation with Prof. J. Roeper (Neurophysiology, Marburg) we study phenotype/genotype correlations of individual DA neurons by combining electrophysiological techniques (in vitro brain-slice patch-clamping) with gene-expression profiling (qualitative and quantitative single cell RT-PCR, DNA-array hybridization) in mechanistically different mouse models of differential dopaminergic degeneration. In addition, we will also analyze related gene-expression in human DA neurons from post mortem brains.
In cooperation with Prof. W.H. Oertel (Department of Neurology, Marburg) and Dr. C Jurinke (Sequenom, US, and within the framework of the NGFN-2, we also analyze genomic single nucleotide polymorphisms (SNPs) in candidate genes to study a possible correlation between genetic variations and PD-risk or disease-onset.
Project Status / Results
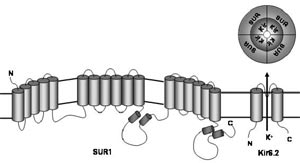
We have made significant progress in understanding the molecular mechanisms of differential vulnerability by investigating the role of a potential downstream target of mitochondrial inhibition and oxidative stress, the ATP-sensitive potassium (K-ATP) channel. This channel is composed of discrete pore-forming (Kir6.1/Kir6.2) and regulatory subunits (SUR1/SUR2) and couples changes in cellular energy metabolism to electrical activity (Fig. 1). Our previous work had demonstrated that these channels are present in dopamine midbrain neurons of young mice, and activated by parkinsonism-inducing toxins like rotenone and MPP+ (Liss et al., 1999). Now we studied the pathophysiological function of these channels in individual identified mesostriatal SN and mesolimbic VTA DA neurons from adult mice under physiological conditions (Fig. 2). For this purpose, we established a novel combination of retrograde tracing, perforated patch-clamping, immunocytochemistry, laser-microdissection, and gene-expression profiling techniques (Liss and Roeper, 2004)
We found that adult SN DA as well as VTA DA neurons possessed functional K-ATP channels with the same molecular make-up (Kir6.2/SUR1). However, in vital in vitro brain slices parkinsonism-inducing toxins activated K-ATP channels and altered electrical activity only in SN DA neurons. In VTA DA neurons, channels were not activated and electrical activity was not altered by inhibition of complex I of the mitochondrial respiratory chain (Liss et al, in press).
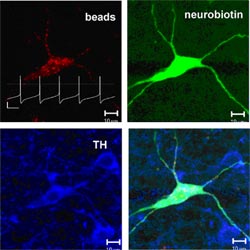
In vivo, this acute functional silencing of SN DA neurons due to K-ATP channel activation would dramatically reduced the release of the neurotransmitter dopamine in the striatum.
To analyze in vivo if selective activation of K-ATP channels is linked to differential vulnerability of DA neurons we analyzed the effect of genetic disruption of the Kir6.2 gene in two chronic and mechanistically distinct mouse models of PD, the MPTP mouse and the mutant weaver mouse. Genetic background controls displayed the typical pattern of selective degeneration of DA neurons. However we observed a selective rescue of the highly vulnerable SN DA neurons in the K-ATP channel knockout mice in both disease models (Liss et al, in press).
These data strongly suggest that the presence and selective activation of K-ATP channels is a death-promoting mechanism, which is necessary for executing the degeneration of dopaminergic neurons. It also provides the first molecular mechanism that might explain the differences in vulnerability to cell-death that occur between different DA populations in PD and its animal models.
Outlook
We currently study upstream control mechanism, that might explain why K-ATP channels are only activated in highly vulnerable SN DA neurons but not in the neighboring VTA DA neurons, that possess also functional K-ATP channels of the same molecular make-up.
In cooperation with our clinical partners, we study the possibility that K-ATP channels act as disease-modifying genes that might influence the age-of-onset or rate-of-progression of Parkinson's disease. To address this, we are currently analyzing genomic DNA of a cohort of early onset PD-Patients and their unaffected siblings for non-synonymous single nucleotide polymorphisms in K-ATP channel genes that would alter the open-probability of the channels. If we find an association between the presence of a specific polymorphism and the manifestation of PD, we plan to further examine these genotyped patients and matched controls. In particular, we plan to perform oral glucose tolerance tests, as the open probability of beta-cell K-ATP channels with the same molecular make-up of those channels in DA neurons is crucial for triggering pancreatic insulin secretion in response to high blood glucose levels.
Lit.: 1. Damier P et al. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain. 1999; 122, 1437-48. 2. Greenamyre JT and Hastings TG. Parkinson's-divergent causes, convergent mechanisms. Science. 2004; 304, 1120-2. 3. Liss B et al. Alternative sulphonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. The EMBO Journal. 1999; 18: 833-846. 4. Liss B and Roeper J. Correlating function and gene expression of individual basal ganglia neurons. Trends in Neurosciences. 2004; 27(8): 475-48. 5. Liss B et al. K-ATP channels promote the differential degeneration of dopaminergic neurons. Nature Neuronscience, in press.