

Introduction
PD is characterized by a loss of dopaminergic neurons in the substantia nigra leading to major clinical and pharmacological abnormalities that are representative for this disease (1). The identification of mutations in different PD associated genes has provided insight into the pathogenesis of this disease (2). In the last years it became obvious that hereditary factors as well as environmental influences have an impact on PD development. Furthermore, PD is a genetically and clinically heterogeneous disorder. Despite these facts it has been postulated that there are common mechanisms underlying PD development. It is assumed that failure of the ubiquitin-mediated protein degradation pathway leading to accumulation and aggregation of proteins may be a common mechanism (3). Alternatively, it has been postulated that the culprit is mitochondrial dysfunction leading to oxidative stress with the possible occurrence of toxic oxidized dopamine species (3). It has been hypothesized that alpha-synuclein, parkin and DJ-1 are involved in these processes.
Project Status
alpha-Synuclein
According to a recent hypothesis by Braak (4) alpha-synuclein pathology spreads from the brain stem and olfactory bulb into cortical areas in a defined pattern. We have subjected samples from human patients representing advanced Braak stages to an investigation of proteinase K-resistant alpha-synuclein in situ and in biochemical extracts. We confirmed that the regional distribution of pathological alpha-synuclein follows the Braak stages, and demonstrated that pathological alpha-synuclein is insoluble, proteinase-resistant, and typically post-translationally modified (5).
We have applied the methodology to our transgenic mouse model for neuronal alpha-synucleinopathy (as it occurs in PD and dementia with Lewy bodies). We found highly susceptible predilection sites in the mouse brain (spinal cord, medullary and pontine reticular formations, deep mesencephalic and subthalamic nuclei) for conversion to pathological alpha-synuclein fibrils (6). These represent a partial, but imperfect overlap with the vulnerable brain areas of human patients. Very recently, we have extended this study to fine analysis of cortical alpha-synucleinopathy in transgenic mice expressing human mutant alpha-synuclein under control of the neuron-specific Thy1 promoter. Interestingly, and highly reminiscent of human patients, the central nucleus of the amygdala was strongly affected by alpha-synuclein pathology in an age-dependent manner. Furthermore, we could establish a temporal correlation of alpha-synuclein misfolding in neurons and behavioural dysfunction in a classical test of rodent amygdala function, namely fear conditioning (manuscript submitted).
First attempts to identify differentially expressed genes that account for this remarkable spatial and temporal specificity of alpha-synuclein misfolding were performed on custom filter arrays, and are currently being refined using Affimetrix chip technology.
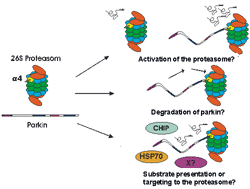
Parkin
The ties between the ubiquitin-proteasome-system and PD were further established by our discovery of the 20S proteasome subunit alpha4 as new parkin interactor protein in a yeast-two-hybrid screen, and our subsequent validation using purified protein preparations as well as cell culture models (7). The significance of this interaction remains to be finally established, and may include a role for parkin in the regulation of proteasomal activity, of its own degradation or of substrate presentation to the proteasome (Fig.1). Preliminary functional data, however, showed that overexpressed parkin could indeed slightly increase proteasomal activity, in line with the first hypothesis (7).
Affymetrix-based expression profiling (testing 7000 known genes) was performed in a PC12 cell system in which parkin overexpression is protective against ceramide induced cell death (8). First looking at an effect of parkin overexpression independent of ceramide treatment, 50 known genes were found to be differentially regulated, 38 up and 12 down (Unschuld et al, resubmitted). Especially low p-values (p<0.001) were found for serotonine receptor 3a (3.4-fold upregulated), the microsomal UDP-glucuronosyltransferase 2B3 precursor (3.0-fold downregulated), integrin alpha M (3.0-fold upregulated) and neuropeptide Y (3.1-fold upregulated). In addition, the array pointed to an upregulation of several genes after ceramide treatment, which was reduced by parkin. The ceramide effect could be validated by real-time PCR vor 4 of 5 tested genes (CHK, EIF4EBP1, GADD45A and PTPN-5) 3 and 6 h after ceramid application, but not 9 or 12 h after ceramid application. Parkin overexpression slightly reduced the upregulation of EIF4EBP1, GADD45A and PTPN-5, but only at 6 h. These results suggest that, in this assay, the cytoprotective effect of parkin might result not only from its E3-ligase activity, but also from direct or indirect modulation of gene expression in a time-dependent manner (Unschuld et al, resubmitted).
We discovered that parkin is phosphorylated (9). Phospho-amino acid analysis revealed that serine was the most prominent acceptor under these conditions. Systematic mutational analysis of serines within consensus sites for common protein kinases pointed to multiple phosphorylated residues in the linker domain as well as in the RING-IBR-RING domain of parkin. We identified several candidate kinases to phosphorylate parkin. Importantly, we could demonstrate that phosphorylation of parkin down-regulated its (auto-) ubiquitin ligase activity, and indeed de-phosphorylation of parkin occurred specifically under ER stress conditions. Thus, we propose that protein-misfolding stress may induce parkin de-phosphorylation and thereby de-repress the ubiquitin ligase parkin. This would facilitate the ubiquitin-proteasome-mediated degradation of misfolded proteins, and explain the neuroprotective mechanism of parkin (9).
DJ-1
Sytuying the the biochemical, cellular and neuropathological features of DJ-1 (10), we have found that [L166P]DJ-1 is highly unstable, which would effectively lead to loss of protein in the affected patients, explain the recessive mode of inheritance in this kindred (11). Another mutation, [E64D]DJ-1 (12) is unremarkable in cell culture, and revealed its deficit only when we investigated the structural dynamics of the purified recombinant protein by circular dichroism spectroscopy (10). [E64D]DJ-1 appeared more rigid compared to [wt]DJ-1, possibly preventing accessibility to a putative active center. Although the exact function of DJ-1 is not known, our finding of up-regulated DJ-1 expression in reactive astrocytes (13) is consistent with the hypothesized anti-oxidative function of DJ-1. We are currently investigating the molecular mechanisms how DJ-1 protects against hydrogen peroxide toxicity, which could be exploited in a (lenti-)viral gene therapy approach.
Outlook
By analyzing simultaneously several PD-related genes using different and complementary methods, we hope to further delineate the interplay and the mechanisms of the different genes/proteins in protecting or aggressing the dopaminergic neurons. This might lead to the identification of new PD candidate genes and ultimatly to new therapeutic approaches.
Lit.: 1. Lang AE and Lozano AM. Parkinson’s disease. N. Engl. J. Med. 1998;339:1130-53. 2. Gasser T. Genetics of Parkinson’s disease. Curr Opin Neurol. 2005;18:363-9. 3. Moore DJ et al. Molecular Pathophysiology Parkinson’s disease. Annu Rev Neurosci. 2005;28:57-87. 4. Braak H et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197-211. 5. Neumann M et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429-39. 6. Neumann M et al. Regional distribution of proteinase K-resistant alpha-synuclein correlates with Lewy body disease stage. J Neuropathol Exp Neurol. 2004;63:1225-35. 7. Dächsel J et al. Parkin interacts with the proteasome subunit alpha4. FEBS Lett. 2005;579:3913-9. 8. Darios F et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Gen 2003; 12: 517-26. 9. Yamamoto A et al. Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J Biol Chem 2005; 280:3390-9. 10. Görner K et al. Differential effects of Parkinson's disease-associated mutations on stability and folding of DJ-1. J Biol Chem. 2004;279:6943-51. 11. Bonifati V et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256-9. 12. Hering K et al. Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7). Hum Mutat. 2004;24:321-9. 13. Neumann M et al. Pathological properties of the Parkinson's disease-associated protein DJ-1 in alpha-synucleinopathies and tauopathies: relevance for multiple system atrophy and Pick's disease. Acta Neuropathol. 2004;107:489-96.