

Introduction
In normal aging and Alzheimer’s disease (AD), Abeta deposition occurs in both parenchymal amyloid plaques and in vessels (cerebral amyloid angiopathy, CAA). However, CAA can also occur in the absence of parenchymal amyloid plaques. For example, patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D) develop a severe form of Aß-CAA but very few amyloid plaques and no neurofibrillary tangles (Levy et al., 1990). These patients suffer recurrent intracerebral hemorrhages leading to death between the ages 45 and 55. HCHWA-D is caused by a point mutation Glu->Gln at codon 693 of the amyloid-beta precursor protein (APP) (amino acid 22 of Aß)(Levy et al 1990).
Although the most common form of cerebral amyloidosis is of the Abeta-type, there are other proteins which have been linked to severe familial forms of cerebral amyloidosis. Hereditary cerebral hemorrhage with amyloidosis Icelandic-type (HCHWA-I), is caused by a point mutation Leu->Gln at position 68 of cystatin C (Ghiso et al., 1986; Levy et al., 1989). HCHWA-I patients suffer the first often fatal hemorrhagic stroke often in their twenties (Olafsson and Grubb, 1999). The amyloid in these patients (ACys) consists of mutated cystatin with an N-terminal truncation of 10 amino acids. Familial British Dementia (FBD) and Familial Danish Dementia (FDD) are autosomal dominant disorders with death occurring at 40-60 years of age (Vidal et al., 1999, 2000). The hallmark lesion in both diseases is cerebral amyloidosis with predominant CAA. In addition the patients develop neurofibrillary tangles (Vidal et al., 1999, 2000). Both diseases are caused by mutations in the recently discovered BRI gene. In FBD a stop codon mutation generates a longer open reading frame while in FDD a 10-nucleotide duplication immediately before the stop codon produces a frame-shift mutation. Both mutations generate 277-amino acid long precursor proteins, ABriPP and ADanPP, that are 11 amino acids longer compared to the 266-amino acid-long wildtype protein (BriPP). In both FBD and FDD, furin-cleavage releases a 34-amino acid carboxy-terminal of the mutant precursor protein to generate the amyloidogenic ABri and ADan peptides, respectively, which are then deposited as amyloid in the brain (Vidal et al., 1999, 2000; Kim et al., 1999).
Although normal aging, AD, HCHWA-D/HCHWA-I, and FDB/FDD share cerebral CAA as common pathology, there are a puzzling variety of additional neuropathological lesions and clinical phenotypes. It is well established that CAA is a risk factor for spontaneous and often fatal hemorrhagic stroke. However, it has also become clear that CAA affects cognition independent of strokes: FBD and FDD patients do not suffer hemorrhage but are demented (Vidal et al., 1999, 2000). Dementia in HCHWA-D is associated with CAA independent of hemorrhagic strokes (Natte et al., 2001). Moreover, CAA in normal aging and AD has recently been linked to cognitive impairment (Pfeifer et al., 2002a) and cerebral hemorrhage following anti-Aß-immunotherapy, one of the currently followed approaches to cure AD (Pfeifer et al., 2002b). Thus, mechanisms by which these amyloidogenic proteins are deposited in the vessel wall and lead to stroke and/or dementia remain puzzling.
Project Status
Over the last five years we have studied APP transgenic mouse models that develop amyloid plaques and CAA. We have also demonstrated that amyloid plaque formation in these mice leads to region-specific neuron death, synaptic and cholinergic dysfunction, and neuroinflammation (Stürchler-Pierrat et al., 1997; Calhoun et al., 1998; Phinney et al., 1999; Stalder et al.,1999; Bondolfi et al., 2002; Boncristiano et al., 2002). Furthermore, we have demonstrated that CAA in these mice leads to a loss of vascular smooth muscle cells, aneurysmal vasodilatation, vessel wall rupture, and recurrent hemorrhages (Calhoun et al., 1999; Winkler et al., 2001; Pfeifer et al., 2002b).
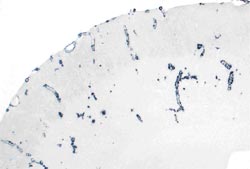
To study the significance of CAA proper, we have developed new APP transgenic mice harboring the HCHWA-D mutation under a neuron-specific Thy-1 promoter (APPDutch mice). These mice develop extensive CAA with only very few parenchymal deposits (Fig. 1). CAA in these mice leads to severe CAA, smooth muscle cell degeneration, cerebral hemorrhages, and neuro-inflammation. In contrast, neuronal overexpression of wild-type human APP (APPwt mice) results in predominantly parenchymal amyloidosis. In HCHWA-D and APPDutch mice the Aß40:42 ratio is significantly higher than in AD and APPwt mice, and significant wild-type human or murine Aß40, respectively, is co-deposited with the AßDutch. Genetically shifting the ratio of AßDutch40:42 towards AßDutch42 by crossing APPDutch mice with mutated presenilin 1 transgenic mice redistributes the amyloid pathology from the vasculature to the parenchyma. The understanding that different Aß species can drive amyloid pathology in different cerebral compartments has implications for current anti-amyloid therapeutic strategies This work has recently been published (Herzig et al., 2004).
To further understand the mechanism by which amyloidogenic proteins are deposited in the vessel wall and lead to stroke and/or dementia, we have also generated cystatin C transgenic mice with the HCHWA-I mutation (Cystatin CL68Q tg mice) and ABriPP transgenic mice with the FBD mutation. For all these transgenic mouse lines the Thy-1 promoter has been used and mice have been generated on a pure B6 background. Unfortunately, the present Cystatin CL68Q tg mice and ABriPP tg mice do not develop cerebral amyloid deposits at least until 24 months of age.
Outlook
The overall aim of our proposal is to elucidate the pathobiological mechanism leading to CAA and to provide a basis for therapeutic strategies. To this end we suggested (a) to generate various transgenic mouse models of familial forms of CAA, and (b) to use state-of-the-art morphological, biochemical, and gene expression and protein profiling to study mechanism how CAA leads to stroke and neurodegeneration.
While such analyses of transgenic mice are ongoing for APPDutch mice, we have started new efforts to generate additional Cystatin C and AbriPP/ADanPP transgenic mice using new refined molecular constructs. To understand the pathomechanism of British and Danish dementia we have also initiated studies aiming at generating Bri-null mice. We anticipate that these new efforts and the current analysis of APPDutch mice will greatly contribute to the understanding of the pathophysiology and therapy of CAA.
Lit.: 1. Levy et al., Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science, 1990, 248: 1124-6. 2. Ghiso et al., Amyloid fibrils in hereditary cerebral hemorrhage with amyloidosis of Icelandic type is a variant of ÿ-trace basic protein (cystatin C). Proc Natl Acad Sci USA, 1986, 83: 2974-8. 3. Levy et al. Stroke in Icelandic patients with hereditary amyloid angiopathy is related to a mutation in the cystatin C gene, an inhibitor of cysteine proteases. J Exp Med, 1989, 69:1771-8. 4. Olafsson and Grubb, Hereditary cystatin C amyloid angiopathy. Amyloid: Int J Exp Clin Invest 1999, 7: 70-79. 5. Vidal et al. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 1999, 399: 776-81. 6. Vidal et al., A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci U S A, 2000 97:4920-5. 7. Kim et al., Furin mediates enhanced production of fibrillogenic ABri peptides in familial British Dementia. Nature Neurosci 1999, 2: 984-88. 8. Pfeifer et al. Cerebral hemorrhage after passive anti-Aß immunotherapy. Science, 2002b, 298: 1379. 9. Sturchler-Pierrat et al., Two APP transgenic mouse models with Alzheimer's disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94: 13287-92. 10. Calhoun et al., Neuron loss in APP transgenic mice. Nature 1998, 395: 755-6. 11. Phinney et al., Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. Journal of Neuroscience 1999 19:8552-8559. 12. Stalder et al. Association of microglia with amyloid plaques in Brains of APP 23 trangenic mice. Am J Pathol, 1999 June 6; 154: 1673-1684. 13. Bondolfi et al. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. Journal of Neuroscience, 2002; 22:515-522. 14. Boncristiano et al. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. Journal of Neuroscience, 2002; 22: 3234-3243. 15. Calhoun et al., Neuronal overexpression of mutant APP results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci, USA, 1999 96:14088-14093. 16. Winkler et al. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. Journal of Neuroscience 2001 21: 1619-1627. 17. Herzig et al. Aß is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nature Neuroscience, 2004, Aug. 15; 7:954-960.