

Introduction
Alzheimer�s disease (AD), like many other neurological and psychiatric diseases presents as a genetically complex disorder. The spectrum of factors that contribute risk to AD is wide and ranges from rare mutations causing an autosomal-dominant early onset form of AD, to susceptibility genes and further to epigenetic and non-genetic risk factors, such as advanced age. Mutations within three genes; the amyloid precursor protein (APP) and the presenilins (PSEN1, PSEN2), account for a large fraction of the familial early-onset forms of AD [1]. Functionally all of these mutations increase the production of the beta-amyloid proteins, consisting mainly of either 40 (A�40) or 42 (A�42) amino acids, which have neurotoxic activity and form the core of the plaques characteristic of AD.
Evidence from twin and family studies supports a significant genetic component to late-onset AD (LOAD), estimating heritability between 60% and 74% [2]. However, LOAD, which accounts for the majority of cases (approx. 95%), shows a more complex pattern of transmission that appears to reflect the action of multiple susceptibility genes. So far only one genetic risk factor for this form of AD has been identified unequivocally; the E4 allele of the apolipoprotein E (APOE) [3].
An oligogenic segregation analysis of LOAD families estimated at least 4 genes contribute to late onset AD, most of which have an effect size equivalent to that of APOE, with one locus estimated to have an effect six fold greater than of APOE [4]. Recent genome-wide linkage studies in patients with late-onset AD have identified several regions with considerable linkage [5-7]. These findings strongly suggest the presence of additional susceptibility genes. One broad region on chromosome 10q (10q21-q24; Multipoint LodScore (MLS) � 4) has gained increasing attention due to the linkage to this region being repeatedly demonstrated in independent patient samples and additionally, by a linkage study using elevated A#42 levels in plasma as a quantitative trait [8-10]. To date the origin of this linkage region has yet to be identified.
The aim of this proposal is the identification of susceptibility genes contributing to the linkage peaks located at the chromosomal regions 6q, 9p, 9q, and 10q.
Results/Project Status
Patient/family recruitment platform
In collaboration with the GSF we established a patient/family recruitment platform based on 20 memory clinics in Bavaria to increase the available numbers of concordant and discordant sib pairs. This network has been systematically extended across Germany and substantially strengthened by press releases, information events, distribution of information material, and the support of the German Alzheimer society. So far, 938 participants have been included compromising 314 sib-pairs/families and 40 extended families with AD.

Establishment of a genotyping platform (TU-Munich)
To ensure the fast and smooth progress of our genotyping activities we entered a cooperation with Sequenom (San Diego, USA) and established a MALDI-TOF based high-throughput genotyping system with a capacity of 50.000 genotypes per day (Autoflex; Sequenom; Fig. 1). Furthermore the close collaboration and integration of the Inst. of Medical Statistics at the TU, which is part of the GEM Munich, facilitates data analyses without any delay.
Identification of genes associated with AD
To decipher the genetic basis of the chromosome 10 linkage peak in AD we applied a combined strategy consisting of a gene-based LD mapping of the LOD-1 region thereby enriching for possible functional candidate-genes. So far we screened over 50 genes in this region and found evidence for a genetic association for a total of four genes. Independent replications are performed using several large case-control samples from Sweden, Australia, the US, and Italy together with a family-based approach using the discordant sib-pair sample. The last step of our validation strategy includes the functional analysis of the gene/polymorphism in collaboration with the other groups of the AD-Subnet. Some of our recent research findings are listed below:
Microsatellite marker D10S1423
Several studies have reported conflicting results concerning the genetic association between AD and the microsatellite marker D10S1423 on chromosome 10p12-14. In an ethnically homogeneous German population of 422 patients with AD and 254 cognitively healthy controls, the 238-bp allele of the D10S1423 marker showed a weak, but after correction for multiple testing no longer significant association with AD (p=0.015, uncorrected; p=0.11, corrected). These findings do not support the presence of a relevant susceptibility locus for AD on chromosome 10p12-14 [11].
Genes involved in cholesterol metabolism
Epidemiological studies identified a higher risk of developing AD among subjects with elevated cholesterol levels. This association may be caused by a modulation of the amyloid precursor protein (APP) processing in response to the cellular cholesterol content. High cholesterol levels may favor the amyloidogenic pathway by inhibition of the alpha-secretase probably leading to elevated beta-Amyloid (Abeta) production. The identification of a linkage peak on chromosome 10q using high Abeta as quantitative trait led us to examine polymorphisms of genes located on chromosome 10 involved in cholesterol metabolism, like Lipase A (LIPA), Cholesterol 25 hydroxylase (CH25H), and FLJ22476, a high density lipoprotein binding related protein. Using 286 patients with AD and 162 controls we analyzed several single nucleotide polymorphisms (SNPs) within LIPA, CH25H, and FLJ22476. None of the polymorphisms showed significant association with AD which contradicts recent findings on CH25H. From our results we conclude that the investigated genetic variations do not contribute to the genetic risk of AD [12].
Choline acetyltransferase (CHAT)
Alterations of the cholinergic system may account for typical clinical and pathophysiological disturbances of AD. In particular, a marked decline of CHAT and as a consequence of acetylcholine during the course of the disease has been described. Due to the chromosomal localization of CHAT at 10q11.23 and its possible role in the pathophysiology of AD, CHAT may represent an appropriate candidate gene conferring risk to AD. In fact, a recent study identified a functional single nucleotide polymorphism (SNP) within the first common exon of CHAT, which was associated with AD giving an odds ratio of 3.8 (Neurosci. Lett. 333 (2002) 9). Because of the potential importance of this finding we analyzed this SNP and another functional SNP within exon 9 (rs868749) of the CHAT gene using a German case control sample consisting of 242 patients with AD and 143 cognitively healthy controls. No statistically significant differences were obtained for the previously described polymorphism. In addition, the exon 9 SNP (rs868749) was not polymorphic in the studied population. We conclude that the previously identified polymorphism is not associated with AD [13].
Insulin degrading enzyme (IDE)
A detailed analysis of the IDE gene locus, through genotyping of 28 SNPs via MALDI-TOF mass spectrometry, was undertaken using a large case-control sample from Germany. Haplotype comparisons with previous studies were based on previously identified haplotype tagging SNPs. Finally, a meta-analysis of IDE haplotype data published thus far, was performed.
Four SNPs (rs11187007, rs2149632_ide12, rs11187033, rs11187040) were found to be associated with AD (p<0.01, uncorrected). Single marker tests with MMSE scores identified significant associations with the most significant result for rs1999763 (p=0.008). Haplotypes reconstructed based on previously described three haplotype-tagging SNPs revealed a significant haplotypic association for the H1 (+H9) haplotype (p=0.028), whilst, H8 with a frequency of about 9% was correlated with higher MMSE scores (p=0.028). A meta-analysis of the major 3-locus haplotypes revealed only the H4 haplotype to have an overall significant effect (pooled OR=1.41 [1.17-1.69 CI]). A subgroup analysis indicated more prominent associations with AD in younger and with MMSE in older patients.
A careful interpretation of the variable locations and different association signals obtained for AD and MMSE may suggest the presence of two independent effects mediated by IDE variants. Firstly, risk for AD and secondly, modification of disease severity. Differences in age structures may contribute to the discrepancies reported so far (submitted)
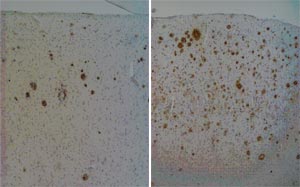
Plasminogen activator urokinase (PLAU):
In a first step we screened a large number of SNPs around the PLAU (~300 kB; 49 SNPs) region using a German case-control sample (AD=448; Ctrls=278) and characterized the LD block structure. We identified a functional SNP (rs2227564 C/T; 141 L/P) at Exon 6 to exert the strongest association, which was located within a region showing low LD (~ 2 kB). To identify any further SNPs which might be responsible for the association we sequenced Exon 6 with the Exon/Intron boundaries at both sides in 80 patients and 80 controls. Only one very rare exonic SNP 1810 C/T; 148 H/Y was identified which was not associated with the disease. The association was then replicated using three independent case-control series from Bonn (AD=109; Ctrls=173), Brescia (AD=120, Ctrls=99), Perth (AD=212, Ctrls=359), and finally one discordant sib-pair sample (251 affecteds; 371 unaffecteds). Possible functional consequences of the PLAU rs2227564 polymorphism on cerebral plaque load was investigated using 33 AD brain tissue samples (submitted).
Prion protein (PRNP)
We investigated the PRNP Met 129 Val polymorphism in 1393 subjects composed of 482 patients with AD and two independent control groups. In patients PRNP Met homozygosity conferred increasing risk with decreasing age at onset (onset: 61-70 years, n=151, p=0.02; OR=1.72 [1.2-2.53]; onset # 60 years, n=138, p=0.013, OR=1.92 [1.31-2.87] whereas no association was obtained in patients with onset above 70 years. The results suggest a stronger involvement of the prion protein into the pathogenesis of EOAD [14].
Tumor necrosis factor alpha (TNFA)
Alzheimer disease (AD), vascular dementia, and stroke are all associated with inflammation though their respective initiating factors differ. Recently a polymorphism in the proinflammatory cytokine tumor necrosis factor (TNF), in association with apolipoprotein E (APOE), was reported to increase AD risk. Two TNFA SNPs, rs1799724 and rs1800629 were genotyped in 506 patients with sporadic AD and in 277 cognitively healthy controls. In a subset of 90 individuals we also investigated whether these SNPs exerted any functional effects on cerebrospinal fluid (CSF) beta-amyloid levels. The frequency of the rs1799724 genotypes and the rs1799724-T allele were significantly different in AD individuals (P=0.009; OR=1.63 [1.13-2.34], while the rs1800629 SNP was not associated with AD. Significant interaction was observed between the rs1799724-T and APOE e4 alleles in that the rs1799724-T allele significantly modified risk associated with possession of the epsilon4 allele only (e4 in absence of rs1799724-T: OR=2.92 [2.00-4.27]; e4 in presence of rs1799724-T: OR=6.65 [3.26-13.55]; P=0.03). Haplotyping analysis revealed a significant overrepresentation of an rs1799724-T/rs1800629-G haplotype in AD (P=0.012; OR=1.60 [1.11-2.29], although to a lesser degree than rs1799724-T alone. Further, the rs1799724-T allele was found to be associated with lower levels of CSF Abeta42 (P=0.023), thus corroborating the genetic findings. Inheritance of the rs1799724-T allele appears to synergistically increase the risk of AD in APOEepsilon4 carriers and is associated with altered CSF Abeta42 levels. Further investigations are warranted to assess the significance of these novel findings [15].
Outlook
The identification of susceptibility genes conferring risk to the AD is the first important step to gain a comprehensive understanding of the aetiology of AD. Only such an understanding will provide the basis of future therapeutic strategies aiming at a sustained modification of the disease progression. Genetic research, in particular the most recent developments allowing whole-genome association studies will undoubtedly have the potential to bring us a significant step forward to reach this goal.
Lit.: 1. Hardy, J., Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci, 1997. 20:154-159. 2. Pedersen, N. et al. Multiple-threshold models for genetic influences on age of onset for Alzheimer's disease: findings in Swedish
twins. Am J Med Genet, 2001. 105:724-728. 3. Corder, E.H., et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science, 1993.261:921-923. 4. Daw, E., et al., The number of trait loci in late-onset Alzheimer's disease. Am J Hum Genet, 2000. 66:196-204. 5. Myers, A., et al., Full genome screen for Alzheimer disease: stage II analysis. Am J Med Genet, 2002. 114:235-244. 6. Blacker, D., et al., Results of a high resolution genome screen of 437 Alzheimer's disease families. Hum Mol Gen, 2003. 12:23-32. 7. Holmans, P., et al., Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet, 2005. 135:24-32. 8. Ertekin-Taner, N., et al., Linkage of plasma Aß42#to a quantitative locus on chromosome 10 in late onset Alzheimer´s disease pedigrees. Science, 2000. 290:2303-2304. 9. Myers, A., et al., Susceptibility locus for Alzheimer´s disease on chromosome 10. Science, 2000. 290:2304-2305. 10. Bertram, L., et al., Evidence for genetic linkage of Alzheimer´s disease to chromosome 10q. Science, 2000. 290:2302-2303. 11. Gohlke, H., et al., Association study between the D10S1423 microsatellite marker and Alzheimer's disease. Neurobiol Aging, 2005. 12. Riemenschneider, M., et al., Association analysis of genes involved in cholesterol metabolism located within the linkage region on chromosome 10 and Alzheimer's disease. Neurobiol Aging, 2004. 25:1305-8. 13. Schwarz, S., et al., Lack of association between a single nucleotide polymorphism within the choline acetyltransferase gene and patients with Alzheimer's disease. Neurosci Lett, 2003. 343:167-170. 14. Riemenschneider, M., et al., Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology, 2004. 63:364-6. 15. Laws, S.M., et al., TNF polymorphisms in Alzheimer disease and functional implications on CSF beta-amyloid levels. Hum Mutat, 2005. 26:29-35.