

Introduction
Parkinson’s disease is characterised by progressive L-dopa-responsive motor impairments including bradykinesia, increased muscle tone, resting tremor, and abnormal postural righting reflexes as originally described by James Parkinson in 1817. The disease is caused by a progressive and selective loss of dopamine producing neurons in the pars compacta of the substantia nigra and, in most cases, by the formation of aggregates called Lewy bodies. The clinical symptoms described above appear after approximately 50% of dopaminergic neurons have lost their functional properties.
However, clinical, neuropathological and genetic experiments revealed, that the term Parkinson’s disease embraces several different degenerative diseases all of which may have different genetic and non-genetic causes. This is also reflected by the great number of genes identified by linkage studies found in large families with autosomal dominant or autosomal recessive inheritance (table 1). Besides the genes involved in monogenic forms of Parkinson’s disease, a growing number of genes termed as “susceptibility genes” exist which, when mutated, increase the risk of the more common sporadic form of Parkinson’s disease. Interestingly, there is increasing evidence that the genes associated with the monogenic forms of Parkinson’s disease may act as susceptibility factors for its sporadic form.
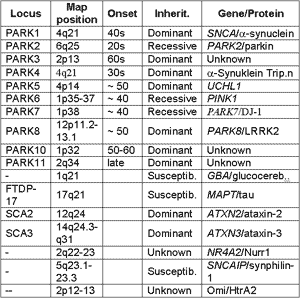
The large number of genes identified not only suggests the existence of more than one "Parkinson’s" disease, but it is also hypothesized that there may be several pathogenetic molecular pathways involved. These pathways may interact and influence each other in order to generate the clinical symptoms used to classify Parkinson’s disease. The findings that genes involved in the aetiology of monogenic Parkinson’s disease are indeed associated and causal for different pathogenetic processes, such as the accumulation of hyperphosphorylated a-synuclein and tau (SNCA/MAPT), the dysfunction of the ubiquitin proteasome pathway (PARK2), the dysfunction of mitochondrial pathways and/or the incapability to compensate for increased cellular oxidative stress (PARK6/PARK7) are supporting this view. To what extend and how these different pathways contribute to the aetiology of Parkinson’s disease is unknown and subject of intense research.
One appropriate tool to tackle this question is the generation of animal models and specifically, the generation of knock-out animals associated with different pathways. In subsequent experiments double- or even triple knock-outs, combining defects in all pathways, should be generated in order to identify the complex molecular interactions leading to Parkinson’s disease. In cooperation with the Universities of Tübingen (Dept. of Genetics, Prof. Riess) and Munich (Dept. of Biochemistry, Dr. Kahle) we are focussing on the establishment of five different models: 1) a knock-out model for parkin, in which only exon12 is deleted; 2) a genetrap knock-out model for DJ-1, 3) a conditional knock-out model for Pink1, 4) a knock-out model for synphilin, and 5) a genetrap knock-out model for periphilin (an interacting partner of synphilin). In our lab animal models for single knock-outs and, later, for double-knock outs are undergoing thorough behavioural and neuropathological studies in order to elucidate the impact of these mutations on neurodegenerative processes and altered dopamine pathways. In our collaborator’s laboratories specific emphasis is put on the in vitro analysis of the protein degradation machinery (ubiquitination, aggregation) and mitochondrial homeostasis (oxidative stress, energy depletion) in these knock-out models.
Project Status
Parkin-knock-out animals
Parkin is encoded by a gene containing 12 exons. Many different mutations, including deletions and multiplications of one or several exons, point mutations altering the open reading frame and thus, producing premature stop codons and truncated proteins as well as changes of a single amino acid in critical regions of the protein, have been identified However, knock-out models, in which the complete protein is absent, have not revealed a function of parkin in the degeneration of dopaminergic neurons in the substantia nigra. Recently, it has been shown that the deletion of the C-terminal part of the parkin protein leads to a truncated form of the parkin protein capable of inducing misfolding. This observation resembles the protein structure of wt-parkin upon oxidative stress, which subsequently leads to the formation of intracellular aggregates. Therefore, we started to generate a mouse model specifically deleting exon12. This is thought to produce the “toxic” truncated form of the parkin protein. Analysis of these mice should reveal whether this truncated protein rather than the absence of the complete protein leads to the degeneration of dopaminergic neurons. Until now, we were able to create chimaeras and these mice are currently breeding for germ line transmission.
DJ-1 knock-out animals
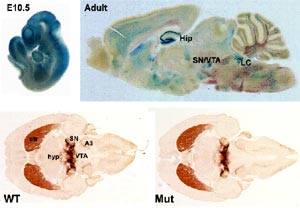
The DJ-1 gene has 8 exons encoding a protein of 189 amino acids that is broadly expressed in mammalian tissues. DJ-1 was initially cloned as a putative oncogene and as part of a multiprotein complex that stabilizes mRNA. Structurally, DJ-1 closely resembles the members of ThiJ/PfpI superfamily, and biochemical studies suggested that DJ-1 might have protease and redox-dependent chaperone activities. Recently, we have shown that DJ-1 can protect cultured cells against the effects of oxidative stress. Different knock-out models, in which the complete protein has been eliminated, were generated. The mutant mice all exhibit subtle phenotypes related to the physiology of the dopamine system. However, the nigrostriatal pathway remained largely unaffected. Nonetheless, DJ-1 appears to have multiple functions potentially relevant to the pathogenesis of PD and it is important to assess how distinct mutations in different parts of the protein affect these functions. We generated a gene trap mutant mouse, missing exon7 and thereby truncating the DJ-1 protein. The first 6 exons of DJ-1 are fused to the ßgeo part of the gene trap vector. These mice are viable and fertile and hence, allowed fine mapping of DJ-1 expression. DJ-1 is ubiquitously expressed during embryonic development and strongly in the adult brain including the hippocampus, amygdala, substantia nigra, locus coeruleus and the nuclei of the cranial nerves in the hindbrain (Fig. 1). LacZ staining of other tissues showed that, as in embryonic development, DJ-1 is expressed strongly in the heart, cortical layers of the kidney, pituitary, gonads and in muscle fibers.
Morphological analysis of 2-month old mice demonstrated no obvious change in the monoaminergic systems in the substantia nigra and the striatum (Fig. 1). However, behavioural analysis revealed that young mice show minor signs of hypokinesia. Further analysis concerning the proteosomal and mitochondrial functionality are in process and preliminary data indicate that the mitochondrial function may be slightly impaired. It remains to be determined whether these subtle defects become more severe in aged animals. This work is performed in close collaboration with subproject 8.2.4 (Dr. Kahle, University of Munich).
Pink1 conditional knock-out animals
Recently, it has been shown that homozygous mutations in the PTEN induced kinase-1 (PINK1) gene causes autosomal recessive young-onset PD. PINK1 encodes a putative protein kinase. These enzymes phosphorylate target proteins, which then perform important cellular roles such as signal transduction. Both, wild-type and mutant PINK1 proteins, primarily locate to the mitochondrion. In cell culture, wild-type PINK1 seems to protect cells from stress-induced mitochondrial dysfunction as well as stress-induced apoptosis, a phenomenon that was abrogated in cells transfected with known human PINK1 gene mutations. Since preliminary expression studies of Pink1 during embryogenesis revealed an ubiquitous expression pattern, we started to generate a conditional knock-out model in which Pink1 is specifically inactivated in dopaminergic neurons. We will intercross a mouse line displaying a floxed Pink1 allele with a mouse line expressing the Cre-recombinase under the DAT-promoter. While Dat-Cre mice are already viable and healthy, floxed Pink1 chimaeras are currently bred for germ line transmission.
Synphilin knock-out animals
Recently, two missense mutations in synphilin, a protein of unknown function but interacting with a-synuclein, have been identified in two sporadic Parkinson patients by our collaborator Prof. Riess (University of Tübingen; subproject 8.2.2). In order to get better insights into the function of this protein a knock-out mouse mutant will be established. In the near future, the knock-out construct will be incorporated into ES-cells and mice will be generated thereof.
Periphilin knock-out animals
In addition, periphilin, a synphilin interacting protein and component of Lewy bodies was recently identified by Prof. Riess (subproject 8.2.2). We generated a gene trap mutant mouse line in which the C-terminal part of the periphilin protein was eliminated. Using this mouse line we could show that periphilin is ubiquitously expressed during embryogenesis and strongly in the adult brain in cortex, striatum and substantia nigra. Interestingly, homozygous mice were not viable and died during early embryogenesis and adult heterozygous did not show any obvious phenotype concerning the morphology of the dopaminergic system.
Outlook
Understanding the molecular signalling pathways leading to the pathogenesis of PD is of pivotal importance for understanding the “many faces” of this disease. The remarkable discovery that multiple genes are pathogenic for Parkinson’s disease implies that more than one disease pathway is responsible for the degeneration of substantia nigra neurons and subsequently, the extrapyramidal motor system. The knowledge about these disease pathways is mostly based on in vitro studies, in vivo models are exceptionally rare. Therefore, the generation of animal models is of uttermost importance to elucidate the role of individual identified genes in the degeneration process of the dopaminergic system in the living organism. However, the task ahead is very challenging because it cannot be expected that the failure of a single gene leads to the full blown pathology of Parkinson’s disease; first analysis of animal models have backed these evaluations. Nevertheless, subtle changes mimicking different aspects of the disease were reliably modelled and combinatory failures of different pathways may lead to the expected pathology. Such an in vivo scenario may be achieved by generating mice deficient for two or three genes known to be associated with different disease pathways in Parkinson’s disease.
The creation of such an in vivo scenario is the aim of our laboratory in collaboration with subprojects 8.2.2 and 8.2.4. Mice deficient of five different Parkinson’s disease associated genes are currently being generated. After a thorough behavioural, morphological and biochemical analysis these mutant mouse lines will then be the basis for intercrosses of mutant mice in which the deleted genes are known to be associated with different disease pathways (i.e. PINK-1 and parkin mutant mice). The analysis of these mice may be the first steps towards a unifying concept in the pathogenesis of PD.
Lit.: 1. Trokovic R et al. Fgfr1-dependent boundary cells between developing mid- and hindbrain. Dev. Biol. 2005 278:428-39. 2. Schnutgen et al. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc Natl Acad Sci USA. 2005 102:7221-7226. 3. Prakash N and Wurst W. Specification of midbrain territory. Cell Tiss Res. 2004 318:5-14.