


Introduction
Epigenetic events, such as deregulated methylation of CpG dinucleotides and aberrant histone acetylation are frequently occurring alterations of the DNA structure that regulate the expression level of proteins. Abnormalities in DNA methylation and histone acetylation have been shown to play an important role in both tumor development and progression thereby impairing the immunogenic potential of cancer. It has recently been shown that DNA hypermethylation and/or histone deacetylation contribute to the lack or deficient expression of components involved in tumor recognition like molecules of the antigen processing and presentation machinery and/or of the costimulatory pathways.
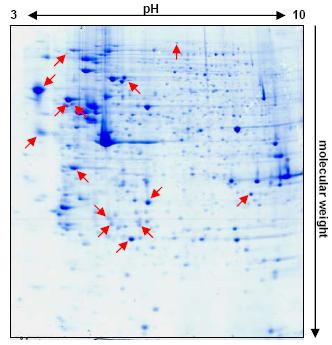
Fig 1: Differences in the protein expression pattern of 5-AZA-CdR treated versus untreated RCC cells. Exclusively expressed or significantly upregulated proteins in the treated variants are marked by red arrows.
Using this approach might not only allow the definition of potential biomarkers, but also might lead to the development of novel immunotherapeutical relevant concepts linked to the reversion of epigenetic phenomena.
Project Status
The group of Barbara Seliger is working since many years on different aspects of the tumor biology and immunology of human RCC. Besides the functional analysis of the HLA class I antigen processing and presentation machinery and of costimulatory pathways in RCC, strategies for the targeting of new biomarkers and therapeutic targets have been investigated using different `ome'-based technologies (Seliger et al., 2002, 2003, 2005; Lichtenfels et al., 2002, 2003; Kellner et al., 2002).
In the course of extensive proteome analyses, the expression profiling of RCC tissues (crude biopsy material/microdissected tissue specimen) and RCC cell lines by 2-DE was standardized and optimised. Protocols for sample preparation, 2-DE and mass spectrometry were established and standard operation protocols (SOP) for each of the processing steps were composed, thus allowing easy sample/data sharing between partners. Furthermore, the PROTEOMEX method which combined proteome analysis with immunoblotting using sera from healthy donors and RCC patients was established and was also demonstrated as an promising approach for the identification of immunoreactive RCC-associated target structures (Seliger et al., 2003; Lichtenfels et al., 2002, 2003).
A large series of well characterized RCC tissue lesions, autologous normal kidney epithelium and corresponding long-term cell lines with a complete history of the relevant clinical data are available as part of the local RCC tumor bank. Moreover, the establishment of cell lines from primary RCC- lesions as well as from corresponding normal renal tissue is well established and optimised in the laboratory leading to success rates in the range of 30-50%. Routine immortalisation with SV40LT and/or telomerase constructs allows the maintaining of respective long-term cell lines.
Both, the RCC tumor bank and the established technology provide the basis for a successful implementation of the present project.
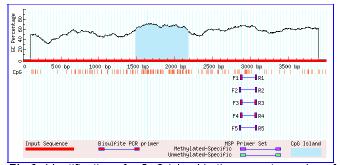
Fig 2: Identification of a CpG island in the promotor region of an epigenetic silencing gene using MethPrimer.
For the identification of cancer-specific epigenetic alterations, different RCC cell lines and normal kidney cell lines are treated with the DNA-methyltransferase (DNMT) inhibitors 5-aza-2`-deoxycytidine (5-AZA-CdR) and 5-aza-cytidine (5-AZA-CR) as well as with the histone deacetylase inhibitor trichostatin A (TSA). Recently, a link has been established between DNA methylation and histone deacetylation by studies showing that DNMT1 and methyl-CpG-binding proteins interact with histone deacetylases and that repression by methylated CpGs is partially relieved by TSA (Rountree et al., 2000; Robertson et al., 2000). Thus we also perform a combined treatment of the demethylating agents and TSA to discover potential synergistically effects on the restoration of the expression of epigenetically silenced genes. Different concentrations of the corresponding agents were tested concerning toxicity effects and apoptosis. In agreement with the literature we finally chose a concentration of 5 �M 5-AZA-CdR, 1 �M 5-AZA-CR and a rather low concentration of 100 nM TSA which show no markedly apoptosis in the selected cell lines. To determine the length of time required to discover a successful treatment on the protein expression level, we performed a time kinetics. After 48h treatment no differences in the protein expression pattern of treated versus untreated cells were detectable in Coomassie Brilliant Blue-stained or silver-stained 2-DE gels. Moreover, immunoblot analyses showed no expression of the cancer testis antigens MAGE-1 and NY-ESO-1 in treated cells, though both were previously identified as epigenetically silenced genes in the corresponding RCC cell lines (Coral et al., 2002).
A contrary result was obtained after 5 days of treatment. Several protein spots were detected exclusively expressed or significantly upregulated in the Coomassie-stained protein expression pattern of the treated variants (Figure 1). Analyses of these spots by matrix assisted laser desorption ionisation-time of flight (MALDI-TOF) resulted in the identification of different proteins, of which some represent interesting candidates for further investigations.
Since gene silencing by hypermethylation is closely connected with the occurrence of CpG island in the promoter region, we examined the promoter regions of the differentially expressed genes using the web program MethPrimer (www.urogene.org/methprimer/index1.html). As expected, several of these genes exhibit CpG islands in their promoter regions indicating that the restoration of their expression is due to the inactivation of promoter methylation (Figure 2).
Recently, the laboratory has established different methods to analyze the DNA methylation pattern of specific genes. These include the COBRA method, a combined bisulfite restriction analysis which circumvents false positives and is even suited for the analysis of a large series of samples. These methods will be used to validate differences in the promoter methylation pattern of treated versus untreated cells.
Outlook
A panel of RCC cell lines representing the major RCC subtypes (clear, chromophilic, chromophobic, oncocytic), metastatic RCC lesions as well as autologous normal kidney epithelium as control will be analysed for their methylation and acetylation pattern. Differences between 5-AZA-CR/5-AZA-CdR and TSA treated and untreated cells identified by conventional 2-DE will be further validated in cooperation with the DKFZ (Heidelberg) by transcriptome analysis using cDNA microarrays. To identify differently expressed proteins with an immunoreactive potential, we will employ the PROTEOMEX technique using sera from tumor patients or healthy volunteers.
In addition, we will further determine the occurrence of differently expressed posttranslational modifications, in particular of arginine methylations which are frequently relevant for the biological function of proteins ranging from signalling, transcriptional activation to protein sorting. For this purpose we will use commercially available arginine methylation-specific antibodies. Analysis of alterations in the arginine methylation of proteins might provide further insights into the mechanisms linked to epigenetic changes in tumor cells.
Our approach to identify and characterise the modulation of the DNA methylation and the histone acetylation status by the use of specific inhibitors might lead to a deeper understanding of the role of epigenetic changes in RCC. Moreover using this approach might allow the identification of novel biomarkers/target structures which will be subsequently validated for their potential implementation to monitor RCC patients during disease and therapeutic treatment and/or the use of these proteins as novel therapeutic targets for the treatment of renal cancer.
Lit.: 1. Seliger et al. Identification of fatty acid binding proteins as markers associated with the initiation and/or progression of renal cell carcinoma. Proteomics 2005. Jul;5(10):2631-40. 2. Seliger et al. Detection of renal cell carcinoma-associated markers via proteome- and other 'ome'-based analyses. Brief Funct Genomic Proteomic. 2003 Oct;2(3):194-212. Review. 3. Seliger et al. dentification of markers for the selection of patients undergoing renal cell carcinoma-specific immunotherapy. Proteomics. 2003 Jun;3(6):979-90. 4. Lichtenfels et al. Identification of metabolic enzymes in renal cell carcinoma utilizing PROTEOMEX analyses. Biochim Biophys Acta. 2003 Mar 21;1646(1-2):21-31. 5. Lichtenfels et al. Heat shock protein expression and anti-heat shock protein reactivity in renal cell carcinoma.Proteomics. 2002 May;2(5):561-70. 6. Kellner et al. Targeting of tumor associated antigens in renal cell carcinoma using proteome-based analysis and their clinical significance.Proteomics. 2002 Dec;2(12):1743-51. 7.Rountree et al. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci.Nat Genet. 2000 Jul;25(3):269-77. 8. Robertson et al. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters.Nat Genet. 2000 Jul;25(3):338-42. 9. Coral et al. 5-aza-2'-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications.Clin Cancer Res. 2002 Aug;8(8):2690-5.