

Introduction
Large-scale association studies have been proposed as a promising means of identifying pre-disposing genes in common complex diseases. Genome-wide linkage disequilibrium (LD) mapping of common diseases could be more powerful than linkage analysis if the appropriate density of polymorphic markers were known and if the genotyping effort and costs could be reduced.
An obvious choice for the analysis of complex diseases are haplotypes, due to their increased informativeness and their potential to condense information on genomic variation. Haplotypes capture information on local LD and on historical recombination and mutation events, thus reflecting the diversity between populations. Haplotypes are also carriers of biological function, both transcriptional and regulatory, which is transmitted as a unit, either from the maternal or the paternal side.
Recent research suggests that discrete blocks of low diversity and high LD exist in the human genome. Within such blocks, information on multiple single nucleotide polymorphisms (SNPs) may be redundant; a non-redundant subset of haplotype tagging SNPs (htSNPs) could be identified and used to distinguish the majority of haplotypes and genomic variation. Genotyping efforts could potentially be simplified by determining an optimal set of htSNPs. This can be done by genotyping a dense SNP map in a small sub-sample of the study population and subsequently restricting genotyping to these htSNPs in the full sample. Such an approach would ensure larger study samples and increased study power without the need for increased funding. Thus, to test a direct effect of a candidate gene or to fine-map an unknown gene by exploiting the pattern of LD, haplotype-based association studies in a case-control design with unrelated individuals or family controls may be more informative and cost-effective than single-marker analyses.
The aims of this project are 1. to clarify basic questions regarding SNP-based association methods for gene mapping in complex diseases, 2. to incorporate the results in new mapping approaches, and 3. to offer recommendations for the application of haplotype-based methods for gene mapping in complex diseases.
Project Status
Haplotype association analysis
The idea of searching for genomic regions shared by cases was first proposed by L. Sandkuijl for genome-wide LD mapping. The haplotype approaches for mapping genes involved in complex diseases are based on the assumption that, in the vicinity of a predisposing mutation, haplotypes carrying this mutation (case haplotypes) are more related to one another than haplotypes not carrying the mutation (random haplotypes). Therefore, the expectation is that the case haplotypes share significantly longer stretches of DNA identical by descent (IBD) around the mutation. The power of gene mapping methods based on haplotype sharing depends strongly on the recombinational and mutational history of the underlying population. Thus, this approach was proposed mainly for gene mapping in homogeneous populations. Houwen et al. (1995) developed a first statistical approach was developed and mapped a new gene for benign recurrent intrahepatic cholestasis, a rare autosomal recessive disease, in a sample that consisted of only three patients in an isolated study population in the Netherlands (1). Haplotype sharing for gene mapping purposes was further developed within the framework of population-based association studies, as well as for use in family-based association studies, and has been applied successfully to both simulated and real data (2).
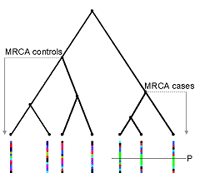
The method presented by Beckmann et al. reflects the flexibility of Mantel statistics using haplotype sharing for gene mapping purposes (2). Mantel proposed a statistic to correlate temporal and spatial distributions of cancer in a generalized regression approach (3). In genetic analyses, spatial similarity is replaced by genetic similarity, and temporal similarity is replaced by phenotypic similarity. The genetic similarity is the shared length between two haplotypes at a marker locus, measured as the length of shared intervals flanked by markers with the same alleles, i.e. by markers that are identical-by-state.
Haplotype block structure
SNP haplotypes combine the advantages of existing genetic markers due to their higher information content when compared to SNPs and to the possibility to define them virtually everywhere in the genome when compared to microsatellites. Haplotypes have raised high hopes for success in association studies for gene mapping and for making genetic studies more efficient. To date, studies with single SNPs have, for the most part, failed for major common complex diseases. Since 2003 the International Human Haplotype Map (HapMap) project is working to catalogue the common haplotypic variation in four different populations genome-wide.
The haplotypic pattern in a region is strongly influenced by the degree of LD between the markers in this region, i.e. the non-random association of their alleles. Since 2001, several publications have suggested the existence of genomic regions that are to some degree independent of their adjacent regions: so-called haplotype blocks. The methods established thus far can be categorized into three groups: 1. focusing on absent recombination events during meiosis in the past generations, 2. looking for regions with sparse numbers of haplotypes, and 3. focusing on high LD in those regions. We developed an entropy-based measure to assess simultaneous LD between multiple markers, which overcomes the limitation of common LD measures for two markers (4). We implemented this measure in an algorithm to define blocks as regions of elevated LD (see fig. 2). An extensive evaluation of this algorithm in collaboration with the Baylor College of Medicine, Houston, part of the HapMap project, proved its usefulness for haplotype marker definition.

Recent work has demonstrated that the block pattern in a particular genomic region depends on 1. the SNP sample density, 2. the considered population, 3. the used block algorithm, and 4. also the SNP selection. This makes the concept of distinct haplotype blocks more complicated than previously imagined. Thus, a future task will be a more comprehensive description of the complex reality of LD structure in the human genome.
Haplotype assignment
In cooperation with Dr Dajun Qian (City of Hope Medical Center, Duarte, USA) we developed a two-stage algorithm to reconstruct haplotypes in population individuals using hierarchical clustering and coalescent tree analysis. Both the similarity between haplotypes as well as the tree distances are calculated based on haplotype sharing. This approach allows for multiple origins of haplotypes within a population. The plausibility of a haplotype is quantified using similar haplotypes within the same cluster, and the effect of random similarities between clusters is excluded. Preliminary results indicate a performance similar to Bayesian approaches. Our approach outperforms alternative methods with respect to accuracy in the presence of missing data.
Tool to simulate data sets
Exhaustive simulation studies are required for the evaluation of statistical methods for haplotype estimation, for the analysis of the haplotype block structure, and for subsequent association analyses. We developed the SNAP software (5), which can generate genotypic data for simulated case-control and nuclear family samples under various LD block designs and disease models. This simulation tool will be used for the evaluation of the methods described below. We added coalescent models to this routine to simulate haplotypes under a number of population history models with differing rates of coalescence, mutation, and recombination. Environmental confounding effects will also be implemented to make use of this software for work planned in the SMP-GEM project on “Statistical methods for gene-environmental interactions”. The simulation tool offers a unified framework for simulating appropriate datasets that resemble complex diseases. It can be used to compare and evaluate different statistical methods for haplotype estimation and association analysis.
Outlook
Haplotype assignment and subsequent association analysis
Any haplotype-based method requires haplotypes corresponding to genotypic data. Techniques for experimentally derived whole-genome haplotypes are becoming available, and such exact haplotypic data are expected to have a significant impact on future gene mapping studies, especially in unrelated individuals. However, the reconstruction of haplotypes from conventional genotype data is still the primary method in most haplotype-based studies, due to the lower cost of genotyping and the availability of fast and accurate haplotyping algorithms. The performance of haplotype-based methods relies on a reliable estimation of the haplotypes. Haplotype reconstruction methods are sensitive to low frequency haplotypes in the range of 1% to 10%, due to sampling error and methodological issues. Genotyping errors and the imputation of missing data also increase the complexity of haplotype reconstruction.
In case-control studies, the question can be raised as to whether haplotypes are jointly estimated for cases and controls, or whether they are estimated separately. Most methods for haplotype estimation tend to make haplotypes within a sample more homogenous. If case and control haplotypes are estimated separately, significant results in a subsequent association analysis might in fact be due to a random process during the haplotype estimation. Conversely, if case and control haplotypes are estimated jointly, it may be more difficult to detect true variants. Differences between case and control haplotypes might be blurred because the estimated haplotypes are more similar than the true ones. Although some authors have proposed methods that take into account case-control status, a comprehensive comparison with respect to subsequent association analysis is lacking. Therefore, we will analyze different approaches to haplotype estimation and subsequent association analysis with respect to: 1. accuracy of haplotype estimation, and 2. the validity and power of the association analysis.
A common approach in haplotype analysis is to use the best estimate of pairs of haplotypes for every individual in the sample. As an alternative approach, we propose Markov Chain Monte Carlo sampling over all compatible haplotype pairs for each genotype according to its probability, as suggested by Rohde and Fürst (6) for samples of unrelated individuals as well as nuclear families. In this approach, not only are the haplotype pair configurations of a study sampled, but in the same step the test for the association is also employed. Tests for association can be performed by the transmission/disequilibrium test TDT for nuclear families, the Mantel statistics for case-control data (2), or an ANOVA for quantitative traits.
Lit.: 1. Houwen R H et al. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat Genet.1994;8:380-386. 2. Beck-mann L et al. Haplotype sharing analysis using mantel statis-tics. Hum.Hered. 2005;59:67-78. 3. Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209-220. 4. Nothnagel M et al. Entropy as a measure of linkage equilibrium over multilocus haplo-type blocks. Hum Hered. 2002;54:186-198. 5. Nothnagel M. Simulation of LD block-structured SNP haplotype data and its use for the analysis of case-control data by supervised learning methods. Am J Hum Genet. 2002;71,A2363. 6. Rohde K et al. Association of genetic traits to estimated hap-lotypes from SNP genotypes using EM algorithm and Markov Chain Monte Carlo techniques. Hum Hered. 2003;56:41-7.