


Introduction
Changes in genomic DNA methylation patterns are an early and consistent hallmark of disease. However, progress in utilisation of this information has been hampered by the unavailability of methods for genomic epigenetic profiling. Because the methylation status of a defined nucleotide can be interpreted to represent a single nucleotide polymorphism, we will utilise microarray technology for a genome-wide, gene-specific analysis of DNA methylation patterns. This approach will eventually result in a detailed characterisation of genomic methylation patterns. The data will be evaluated together with the available clinical data and results from transcriptional profiling. They will allow fundamental insights into the role of DNA methylation during disease development and will provide the foundation for an epigenetic classification of disease.
The group assembled in this platform has established the means for (1) the sodium bisulfite modification of DNA samples for the generation of methylation-dependent polymorphisms, (2) the detection of the resulting single-base polymorphisms on complex oligonucleotide microarrays and (3) bioinformatics platforms for subsequent data analysis, with a special focus on combining the epigenetic information with transcript profiles and clinical information. Also, for the analyses performed within the SMP, clinical material with corresponding clinicopathological information is available.
As a group, we will provide a platform for screening for new, informative epigenetic markers for relevant diseases. In addition, well-established markers will be put to use. Apart from the provision of biological data, we will concomitantly advance and thus improve various technological aspects. Infrastructure will be provided that will be used in collaborations within the NGFN-2 network and beyond for a global analysis of epigenetic variations by means of microarray-based analyses. Different systems, which meet the respective requirements, will be provided. By comparison between them, standards will be defined that will allow comparability of data across platforms and across various areas of analysis. Also for this purpose, the existing close connection and interrelation of this partnership with international efforts is crucial.
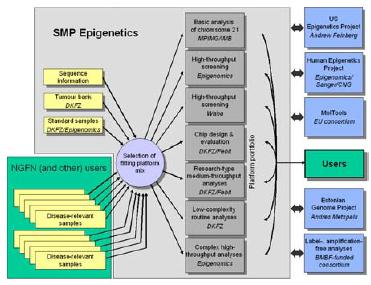
Fig 1: Overall structure of the SMP Epigenetics
In the human genome, about 4% of the cytosine residues are modified by methylation at their carbon-5 position. DNA methylation plays a key role in genomic imprinting and the maintenance of genome integrity by transcriptional silencing of repetitive DNA sequences and endogenous transposons, thus preserving chromosome conformation and preventing aberrant homologous recombination events. Last, DNA methylation represents an epigenetic mark that regulates the expression of a large number of genes. This regulation involves a variety of factors, like DNA methyltransferases, methyl-DNA binding proteins and chromatin proteins. The synergistic action of all factors involved results in the establishment and maintenance of a specific DNA methylation pattern. This genomic methylation pattern represents the epigenetic programme of the genome.
Epigenetic programming of the genome dictates the interpretation of the genetic information. For example, different cell types can be distinguished by their cell type specific epigenetic programmes. Similarly, tumour cells, for example, can be distinguished from normal cells by a tumour-specific epigenetic programme. These differences in epigenetic programming cause concomitant differences in gene expression patterns. Aberrant methylation patterns are responsible for or at least associated with many common diseases. However, most analyses carried out to date are restricted to single loci. In addition, detailed information on differences between healthy and disease tissue is frequently non-existing, although it could be used not only for diagnostic purposes but also prognosis of disease progression or even the development of therapy.
Various methods exist to analyse the variation of cytosine methylation. However, only the introduction of the sodium bisulfite reaction, which converts unmethylated cytosine to uracil while methylated cytosine remains unaffected, provided the means for more global studies. The chemical reaction produces methylation-dependent single nucleotide polymorphisms (SNPs), which can be assayed by appropriate processes. For global analyses, the use of microarrays currently offers on of the best options. Very many sites can be analysed in parallel and in a (semi-) quantitative manner. Also, many identical microarray copies can be generated, thus permitting the analysis of a large number of different samples.
Subproject No. 8.3: Analysis of DNA methylation patterns by oligonucleotide extension arrays.
This subproject will focus on establishing a flexible analytical system that fits the particularly high complexity of mammalian DNA methylation patterns. In previous experiments – part of collaboration between Frank Lyko and Jörg Hoheisel, funded by the DFG, and as part of a project between Jörg Hoheisel and Epigenomics in NGFN-1 – we started the assembly of a database containing methylatable DNA sequences from cancer-associated loci. In addition, we have established the sodium bisulfite deamination protocol for genomic DNA samples provided by clinical collaboration partners from independent projects. The sodium bisulfite procedure generates methylation-dependent single nucleotide polymorphisms on the DNA that can subsequently be detected on complex oligonucleotide microarrays by primer extension reactions. The strategy relies on the use of a polymerase for discrimination, since the enzymatic specificity of base-calling is by a factor of 10- to 100-fold better than the detection of mere differences in duplex stability. The oligonucleotides are designed to fit the sequence directly adjacent to the polymorphism site. If a dideoxynucleotide is added, extension of the oligomer occurs only if the base was added that is complementary to the nucleotide present in the annealed DNA-molecule at the position of polymorphism. The detection of cytosine methylation is of reduced complexity, since in either strand only incorporation of one of two nucleotides can take place. In addition to improved sensitivity by the polymerase reaction, the selection of the oligonucleotide sequences is simplified, since the respective duplex stability is much less critical. Also, the throughput of the process can be multiplexed much better, since the assay is less susceptible to falsely binding target molecules. Additionally to the degree of complementarity between probe and target sequence, the polymerase will distinguish between correct and incorrect templates.
In an existing cooperation with the company Febit biotech (Heidelberg), we tested their flexible array platform. It allows an entirely flexible design of each individual microarray and provides low cost per oligomer – due to light-controlled but mask-free in situ synthesis – and sufficient capacity with currently up to 48,000 oligomers per array, split into eight independent channels. At the same time, we developed a synthesis chemistry that conforms to biological synthesis direction: the resulting oligomers are attached via their 5’-ends, while the 3’-termini are freely accessible. Additionally, synthesis yields are near quantitative, so that also long oligomers can be synthesised in sufficient quantity (Beier et al., 2001). Sites to be analysed in our subproject will be selected from public databases and also include loci identified in the genome-wide screen on CpG-island microarrays performed in the subproject by Andreas Waha. In addition, critical genes identified by the partners within NGFN-2 or beyond will be searched for sequences, which are conserved between mouse and human and/or likely to be methylated, as determined by their CpG content. Any potential methylation site within such a sequence will be also placed on the chip and assessed by experimentation. The parallel fluidic system of Febit’s Geniom array system consists of currently 8 channels, which can be used independently for analysis, if required. For most polymorphic sites, one oligomer will be synthesised for each strand. Since each channel holds 6,000 oligomers, 3,000 different methylation sites could thus be analysed in each channel, actually well fitting to the degree of multiplexing that could be expected with to the polymerase extension reaction. Using all channels simultaneously, up to 32,000 analyses could eventually be done simultaneously in total on a single chip.
Based on the combination of the Febit hardware and our technology for the synthesis of oligonucleotides, we intend to analyse the variation in methylation of several cancer entities. Due to the freedom in the length of the initial oligomers, synthesis of 50-70mers is possible, rather high temperatures can be applied. Consequently, longer fragments of DNA can be used in the hybridisation, reducing problems caused by intramolecular folding. Andres Metspalu and co-workers at the Estonian Biocenter, with whom we collaborate on the use of primer extension reactions for genotyping, have analysed many samples simultaneously in a single reaction on arrays made of spotted oligonucleotides (personal communication). Their protocols were transferred to our laboratories and will be implemented on the Febit system. Both the degree of extension and the percentage of extended oligomers can be increased by a cycling reaction. Since the Febit chip is a contained system, no evaporation is taking place. With about 3 µl of liquid in each channel, cycling can be performed rather quickly. Also, the reaction could be cycled with decreasing levels of hybridisation stringency, as long as the individual results per cycle can be monitored (as is the case with the process proposed here). Thereby, very specific signals would be recorded first before stringency is reduced for the generation of more information of less overall accuracy.
The relevant tissues are available from the tumour bank organised by Prof. Gröne as well as other partners. Pools of selected PCR-products will be made from the genomic regions of interest. The results obtained on the chip will partly be validated by independent established methods, such as methylation-specific PCR and bisulfite sequencing. These methods are of lesser throughput but proven in terms of their performance. Parameters such as DNA concentration, total reaction volume, kinetic factors and nucleotide concentrations during the polymerase reaction will be optimised. Initial results exist already from the DFG-funded project (Hoheisel/Lyko). Complex oligomer arrays will be produced on this basis in order to check their performance. Due to the flexibility of the chip-design, there will be a continuous development of the arrays. Nevertheless, detailed epigenetic analyses will be performed right from the start. Only the throughput will increase steadily during the project, since more and more well-characterised oligomer probes will become available.
Simultaneously, it is planned to compare the data on epigenetic variations with information on transcriptional variations and differential splicing gained within the SMP RNA Profiling. In addition, some parallel analyses will be conducted on the actual level of protein expression in a project funded within the BMBF Proteome Network. Potential correspondence between epigenetic variations and changes at any such level will be analysed by a software programme, which co-localises by a special cluster algorithm in one plot corresponding factors from different types of analysis (Fellenberg et al., 2002; Busold et al., 2005). Lastly, the clinical data available from our cooperation partners for every patient will be included. Biostatistical analysis of this data has provided intriguing insights into the function of DNA methylation and also allows the establishment of novel diagnostic tools based on DNA methylation analysis.
Lit.: Beier, M., Stephan, A. and Hoheisel, J.D. (2001). Synthesis of photolabile 5'-O-phosphor-amidites for the production of microarrays of inversely oriented oligonucleotides. Hel. Chim. Acta 84, 2089-2095. 2. Fellenberg, K., Hauser, N.C., Brors, B., Hoheisel, J.D. & Vingron, M. (2002). Microarray data warehouse allowing for the statistical analysis of experiment annotations. Bioinformatics 18, 423-433. 3. Busold, C., Winter, S., Hauser, N., Bauer, A., Dippon, J., Hoheisel, J.D. & Fellenberg, K. (2005). GO-annotations in correspondence analysis facilitate interpretation of microarray data. Bioinformatics 21, 2424-2429. 4. Mund, C., Beier, V., Bewerunge, P., Dahms, M., Lyko, F. & Hoheisel, J.D. (2005). Array-based analysis of genomic DNA methylation patterns of the tumour suppressor gene p16 promoter in colon carcinoma cell lines. Nucleic Acids Res. 33, e73.