

Introduction
In cross-sectional epidemiological studies, a random sample is usually drawn from the general population in to make conclusions about the underlying general population. This approach assumes that the response probability is the same for all subjects in the population. However, it is known (1) that participants and non-participants differ in terms of their personal and disease-related characteristics. Such variation in response liability may impede the accuracy of a study and may bias any estimates obtained from it. This important problem has not hitherto received adequate attention, particularly not in genetic studies. In survey-based studies, characteristics of non-participants can be derived from information obtained during the contacting process or through a short interview. It has been proposed that participants who enter the study comparatively late show similar characteristics to non-participants (2). This hypothesis appears sensible since late participants would have turned non-participants if the contact period had been shorter or the effort had been less intense.
However, whether investigators have to account for a response bias in genetic association studies, because of different allele frequencies in participants and non-participants, is as yet unclear.
In genetic association studies between genetic variations and complex diseases, the problem of an inhomogeneous population with respect to allele frequencies is known as population stratification. Population stratification occurs, when the population of interest consists of subgroups, that have different allele frequencies for a gene on the chromosomal region of interest. If these subgroups also have different frequencies of a true risk factor, then subgroup membership is a confounder. In recent years, only a small fraction of significant association results has been replicated by other studies. Undetected genetic substructures in the population may be one of the reasons for spurious or biased results.
Thus, this project is aimed at assessing the genetic heterogeneity between early and late participants in the S4 Survey (1999/2001) of the KORA study (Cooperative Health Research in the Region of Augsburg), in order to estimate the possible bias introduced into genetic association studies by low response rates. Other factors as age, origin of the parents and the urban-rural difference are also considered as confounding factors.
Results/Project Status
Non-response as a risk factor in population-based studies
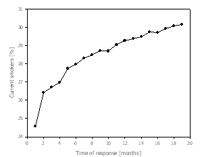
In the MONICA/KORA studies in Augsburg, we have found that non-response is a relevant disease risk factor. When mortality-follow-up was performed in a cohort of 6115 individuals recruited from the general population, 1093 were found to be non-participants. After a follow-up period of 8.3 years, mortality among participants was 460/100.000 person years as opposed to 752/100.000 person years among non-participants (RR=1.7). Furthermore, when we analysed data from KORA survey S4 for time of participation, we found that smoking was prevalent in 27% of early participants, and in 39% of late participants (see Figure 1). The prevalence of type 2 diabetes was 3.7% in early participants and 5.2 % in late participants (3). Here, early and late participants have been defined by a response time from the first invitation to participation of less and more than 3 months respectively.
Thus, there is a clear indication, that response time correlates with risk factors for many complex diseases in the KORA study, that are also subject to genetic research. If genetic heterogeneity between responder subgroups can be detected, response time is found to be a confounding factor. Particularly in view of the positive association between smoking and response time observed in KORA, it will be highly interesting to investigate SNPs in the recently proposed addiction genes (4). Since blood samples from the KORA S4 survey are used as controls for different case groups for case-control studies – either from the same population or other populations – the possible population stratification is also of general interest (5).
Population stratification between three German populations
A study has been performed to investigate whether genetic differences exist between the KORA S4 survey, representing a Southern German population, and two Northern German populations (PopGen in Kiel and SHIP in Greifswald). A total of approximately 2100 individuals from these three areas were included, spanning an age range of 25 to 74 years. Individuals were genotyped for 206 autosomal markers from coding regions as well as from the “genomic desert”. All SNPs are uniformly dispersed throughout the genome and have moderate allele frequencies. After the selection process, these SNP loci have been used for the Genomic Control (6) and Structured Association (7) methods. Both methods are based on the idea, that population substructure not only affects the candidate genes, but also other genes. Alleles that are not associated with the disease or candidate genes can be used to assess the existence of population substructure in a sample and identify the underlying subgroups (8). First results showed, that there is non-negligable genetic heterogeneity between the populations according to the geographical distance (9). Although significant population substructures could be found by calculating the inflation factor #, the model-based clustering approach proposed by Pritchard et al. (7) and implemented in the programme STRUCTURE could not detect relevant population structures. In the situation of subtle population stratification, however, as it is expected in this case, this method has been demonstrated to show limited sensitivity [10].
Population stratification within KORA
The programme STRUCTURE has also been used to take a closer look at population stratification within the KORA sample. The 206 SNPs that have been genotyped in the KORA S4 subpopulation (n=730) have been used to infer the presence of distinct populations and assign individuals to one of a number of populations (K), that is a priori defined. For the KORA sample, the posterior probability for K=1 was approximately 1, leaving 0 probability for the alternatives, that this very sample consists of more than one population. Figure 2 shows, that under the assumption of three populations, all individuals are assigned the same probability for all clusters and cannot be allocated into one of these.
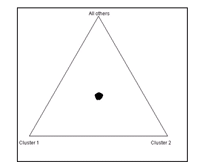
This doesn’t exclude the possibility, though, that there is a subtle population stratification, that can still lead to a bias in the association estimates when not accounted for.
Within the KORA sample, the admixture rate alpha was estimated to be 3.82. Since rates of more than 1 indicate a high extent of admixture [11], most individuals in this sample are presumably admixed out of several subpopulations.
Outlook
Our first results show, that late participation is a risk factor in epidemiological studies and can therefore also be the cause for population stratification and thus leading to false-positive results in genetic association studies. Significant population stratification has been detected between geographically differentiated populations within Germany. However, separate populations couldn’t be found within the KORA subsample. The role of late participation and the non-response mechanism remains to be investigated. The methods of Genomic Control used in the comparative German Genomic Control study, will be applied to test the genetic differentiation between responder subgroups. To answer the question of genetic homogeneity as general as possible, focus has to be upon candidate genes for multiple complex diseases and metabolic pathways, as well as on non-coding drift-sensitive null-loci. Since no DNA is available from non-participants, the analysis will have to be confined to comparing early and late participants. However, data on participants and non-participants can be simulated assuming the underlying distribution. A broad variety of assumptions regarding the participant-specificity of allele frequency distribution can thereby be tested in order to assess the magnitude of possible bias.
In a recent Scottish study [12], genetic differentiation was tested between one urban and nine rural regions, showing a clear difference in LD patterns between these regions. To investigate this source of population stratification in our study, the analysis will be extended to include the residential community of each individual.
The possible genetic differentiation introduced by the origin of the individuals’ parents will also be a focus of further investigations.
Lit.: 1. Schnell R, Nonresponse in Bevölkerungsumfragen. Ausmaß, Entwicklung und Ursachen. Opladen 1998 2. Richiardi L, Boffetta P, Merletti F. Analysis of nonresponse bias on a population-based case-control study on lung cancer, Journal of Clinical Epidemiology. 2002; 55: 1033-1040 3. Hochadel M, Holle R, Wichmann H.-E Distribution of risk factors and disease prevalences depending on early or late response, submitted 4. Lerman C, Berrettini W. Elucidating the role of genetic factors in smoking behavior and nicotine dependence, Am J Med Genet. 2003; 118B(1):48-54 5. Wichmann H.E., Gieger C., Illig T., for the KORA Study Group. KORA-gen - Resource for population genetics, controls and a broad spectrum of disease phenotypes. KORA-gen - Ressource für Bevölkerungsgenetik, Kontrolle und ein breites Spektrum an Krankheitsphänotypen. Gesundheitswesen 2005; 67. 6. Devlin B, Roeder K. Genomic control for association studies. Biometrics 1999; 55(4):997-1004 7. Pritchard JK, Donnelly P. Case-control studies of association in structured or admixed populations. Theor Popul Biol 2001; 60(3):227-237. 8. Lamina C., Steffens M., Mueller J., Lohmussaar E., Meitinger T., Wichmann H.-E. Genetic diversity in German and European populations: Looking for substructures and genetic patterns. Genetische Diversität in deutschen und europäischen Bevölkerungen: Suche nach Substrukturen und genetischen Mustern. Gesundheitswesen 2005; 67. 9. Steffens M. et al. SNP-based Analysis of Genetic Substructure in the German Population, in preparation 10. Hao K, Li C, Rosenow C, Wong WH. Detect and adjust for population stratification in population-based association study using genomic control markers: an application of Affymetrix Genechip Human Mapping 10K array. Eur J Hum Genet. 2004 Dec;12(12):1001-6. 11. Pritchard JK, Stephens M, Donnelly P..Inference of population structure using multilocus genotype data. Genetics. 2000 Jun;155(2):945-59. 12. Vitart V, Carothers A, Hayward C, Teague P, Hastie ND, Campbell H, Wright AF. Increased level of linkage disequilibrium in rural compared with urban communities: a factor to consider in association-study design. Am J Hum Genet. 2005 May;76(5):763-72.