

Introduction
Vogelstein and Kinzler summarize recently that alterations in three types of genes are responsible for tumorigenesis: oncogenes, tumor suppressor genes and stability genes. While oncogenes are mutated in ways rendering the gene constitutively active, tumor suppressor genes are targeted in the opposite way by reducing the activity of the gene product. The outcome of either active oncogenes or inactive tumor suppressors converge to the similar path: They drive the neoplastic process by increasing tumor cell numbers through the stimulation of cell proliferation or the inhibition of cell death or cell cycle arrest. Thus, beside other important features, the uncontrolled cell cycle is one of the most striking hallmarks in human tumors, leading to progressive cell proliferation.
In recent years the regulation of the cell cycle has been extensively investigated. In mammalian cells, the cell cycle is under temporal and spatial control by a family of protein kinases, the Cdks and their regulatory subunits, the cyclins. Regulation of Cdk activity occurs at multiple levels, including cyclin synthesis and degradation, phosphorylation and dephosporylation, Cdk inhibitor (CKI) protein synthesis, binding and degradation, and subcellular localization. The activation also requires the Cdk-activating kinase (CAK) that phosphorylates Cdk subunits at residues Thr 160/161, and the Cdc25 phosphatase that dephosphorylates residues Thr 14 and Tyr 15. On the contrary, most types of cancer display a deregulated cell cycle. This can be a consequence of either the aberrant expression of positive regulators, such as the cyclins, or the loss of negative regulators, like the CKIs. The components that are intensively studied and implicated in transformation are mostly involved in the regulation of G1 and S phase or DNA damage checkpoints including the pRB- and p53 pathways. However, data emerging in recent years demonstrate that in addition to the alterations in G1 and S phase, a defective G2/M transition is also associated with oncogenesis. Among many G2/M regulators we have focused on two molecules responsible for initiation and passage through the mitosis, namely Plk1 and cyclin B1, the regulatory subunit of Cdk1 (Cdc2) (Yuan et al., 2002a; Eckerdt et al., 2005a; Eckerdt et al., 2005b; Yuan and Strebhardt, 2005).
Named after the polo gene of Drosophila melanogaster, Plks that code for serine/threonine kinases, constitute a novel, evolutionarily conserved family of essential cell cycle regulators. Mammals express at least three different Plks: Plk1, Plk2 (Snk) and Plk3 (Fnk/Prk). Among them, human Plk1, which was identified and cloned by us (Holtrich et al., 1994), clearly functions during different mitotic phases and is most likely to represent the functional homologue of Drosophila Polo. Plk1 is a cell cycle-regulated kinase in context of its protein level, activity and localization. As cells progress through the cell cycle, Plk1 protein level is low in G1, accumulates during S and G2, reaches its maximal level as cells approach M phase, and is rapidly reduced after mitosis. During mitosis, Plk1 activity peaks, concomitant with maximal Plk1 phosphorylation. Furthermore, Plk1 undergoes transient associations with both the spindle apparatus and the kinetochore/centromere region of mitotic chromosomes. Finally at the exit of mitosis, Plk1 is proteolytically degraded through the ubiquitin-proteasome pathway.
Shortly after the cloning of first member of the Plk family, polo, human cyclin B1, was identified. The human B-type class of cyclins consists of three closely related members, cyclins B1, B2, and B3, which control the G2/M transition through combination with the Cdk protein kinases. Among these, cyclin B1 is the best studied and characterized member of the cyclin B-family. Activation of the Cdk1 kinase only occurs when sufficient cyclin B1 protein has been synthesized. The expression of the cyclin B1 protein correlates with mRNA accumulation. Human cyclin B1 mRNA appears at the end of S phase and reaches its peak during G2 and M phases. Like Plk1, the proper regulation of cyclin B1 is critical for the entry into mitosis.
As shown in Fig. 1, both, cyclin B1 and Plk1, are deregulated and overexpressed in many tumor cells and contribute greatly to the progressive proliferation of tumor cells. Moreover, the overexpression of Plk1 or cyclin B1 can override cell cycle checkpoints and drives cells into mitosis regardless of cellular damage. Consequently, the deregulation of both kinases is connected with chromosome instability characteristic to tumoral development. Finally, the high expression of cyclin B1 or Plk1 is observed in a variety of primary tumor tissues, serves as a proliferation marker (Yuan et al., 1997) and a poor prognostic marker for tumor patients, and very often associated with the resistance to chemotherapy or radiotherapy. Thus, targeting these key players of the G2/M transition could represent a novel promising strategy for anticancer therapy (Eckerdt et al., 2005b; Yuan and Strebhardt, 2005).
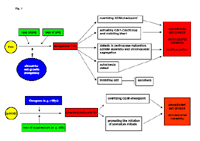
Fig 1: Schematic illustration of the involvement of Plk1 and cyclin B1 in tumorigenesis.
Results
Plk1 as a therapeutic target
To investigate the therapeutic potential of targeting Plk1, phosphorothioate antisense oligonucleotides (ASOs) were firstly applied to selectively downregulate Plk1 expression in MDA-MB-435 (breast cancer), HeLa S3 (cervical carcinoma) and A549 (non-small cell lung cancer) cells (Spänkuch-Schmitt et al., 2002a). Specific ASOs were identified which suppress Plk1 mRNA as well as protein in a dose-dependent and sequence-specific manner. Moreover, cells treated with Plk1 ASOs displayed a rounded phenotype with multiple centrosomes. Furthermore, ASO treatment had a potent antiproliferative effect in cell culture as well as in xenografted nude mice based on A549 cells. These studies suggest for the first time that the reduction of Plk1 inhibits strongly the proliferation of human tumor cells. Thus, ASO inhibitors targeting Plk1 at well-tolerated doses may be considered as potential anticancer agents.
In a RNA interference-based approach small interfering RNAs (siRNAs) were applied to deplete Plk1 in tumor cells (Spänkuch-Schmitt et al., 2002b). The results showed that siRNA is more potent and specific than ASOs in the context of reducing Plk1 protein level and of inducing an antiproliferative effect on human tumor cells, suggesting that siRNAs targeted against Plk1 could be valuable tools for cancer therapeutic intervention by displaying its activity against a broad spectrum of neoplastic cells at very low dosages. More importantly, we could further demonstrate that cancer growth was inhibited in xenograft mice after systemic application of the promoter-driven short hairpin RNAs against Plk1, in combination with the nuclease inhibitor aurintricarboxylic acid (ATA) to extend the lifespan of the plasmids in murine blood (Spänkuch-Schmitt et al., 2004).
In addition to ASOs and siRNA-based techniques, membrane-permeable inhibitory peptides were also designed to reduce the activity of Plk1 (Yuan et al., 2002b). The C-terminal regions of Plks contain conserved domains termed polo-boxes, which are required for subcellular localization and for physical interaction with substrates. We linked the most conserved 20 amino acids (aa 410-429) of the polo-box domain to an antennapedia peptide and studied its antiproliferative effect on different human tumor cell lines. While a mutated derivative was much less effective, the wild-type polo-box peptide inhibited the proliferation of tumor cells associated with induction of apoptosis and G2/M arrest. The treatment with wild-type polo-box peptides also caused misaligned and multiple centrosomes.
Collectively, studies based on different techniques demonstrate that Plk1 is a promising target for antitumor therapy. The depletion/reduction of Plk1 resulted strongly in an inhibition of proliferation of tumor cells in vitro and in vivo.
Cyclin B1 as a therapeutic target
We analyzed recently the impact of siRNA against cyclin B1 on the proliferation of different human tumor cell lines (Yuan et al., 2004). We could show that cyclin B1 siRNA reduced specifically and efficiently the protein level of cyclin B1, consequently also the kinase activity of Cdk1/cyclin B1 in various tumor cell lines. In contrast to primary human umbilical vein endothelial cells, the proliferation of treated tumor cells was strongly inhibited by arresting cells at G2/M phase and further inducing apoptosis. Moreover, tumor cells displayed an impaired colony forming ability after siRNA treatment. These data indicate that the downregulation of cyclin B1 could become a specific strategy for the inhibition of Cdk1/cyclin B1 as an antiproliferative intervention, once the hurdles of siRNA technique are overcome in the future. Alternatively, small molecular compounds against specific domains of cyclin B1 could be more attractive, either blocking the functional binding of cyclin B1 to Cdk1, or inhibiting the translocation of Cdk1/cyclin B1 complex from cytoplasm to nucleus. It is tempting to suggest that the acceleration of the degradation of cyclin B1 protein could be also an interesting intervention by triggering the proteasome-mediated ubiquitination of cyclin B1. In the light of recent studies published and our own unpublished data, we are convinced that beside promising small molecular compounds targeting Cdk1, the abrogation of cyclin B1 function could be a more specific strategy to inhibit the activity of Cdk1, stopping the progressive train of cell division and inducing apoptosis in tumor cells.
Taken together, our studies demonstrate that downregulation of Plk1 or cyclin B1 inhibits specifically the proliferation by arresting cells at G2/M phase and by inducing apoptosis in tumor cells, whereas normal cells exhibit no apoptosis and a much less extent of G2/M arrest.
Outlook
We are going to translate the results from NGFN1 to pre-clinical trials by exploiting new compounds against Plk1 and cyclin B1.
(1). To finish pre-clinical trials with Plk1 antisense oligos in breast cancer cells as well as in mouse model in combination with Herceptin (monoclonal antibody against HER2 receptor).
(2). To validate the results of the small inhibitory polo-box peptides targeting Plk1 in animal models and to accomplish pre-clinical studies of this agent.
(3). To search for small molecular weight compounds against Plk1 and cyclin B1, mainly in collaboration with our partners in NGFN2 (Dr. R. Frank, GBF, Braunschweig; Dr. T. Berg, MPI, Martinsried).
With the expanding knowledge of cell cycle regulation in tumor cells, our studies by blocking G2/M transition and inducing apoptosis of tumor cells may contribute to future tailored treatment for tumor patients. The further search for combinations with other suitable targets or conventional chemotherapy could open a new door in context of intervention and treatment of human cancer.
Lit.: 1. Eckerdt, F., Yuan, J., Saxena, K., Martin, B., Kappel, S., Lindenau, C., Kramer, A., Naumann, S., Daum, S., Fischer, G., Dikic, I., Kaufmann, M & Strebhardt, K (2005a) Polo-like kinase 1 (Plk1) mediated phosphorylation of Pin 1 stabilizes Pin1 by inhibiting its ubiquitination in human cells. J. Biol. Chem. Aug 23 (Epub ahead of print). 2. Eckerdt, F., Yuan, J. & Strebhardt, K (2005b) Plk and Oncogenesis. Oncogene 24(2), 267-276. 3. Yuan, J. & Strebhardt, K (2005) Targeting the G2/M transition of the cell cycle for tumor therapy. Letters in drug design and discovery 2, 274-281. 4. Spänkuch-Schmitt, B, Mattheß, Y., Zimmer, B., Kaufmann, M. & Strebhardt, K (2004) Cancer inhibition in nude mice after systemic application of U6 promoter-driven siRNAs targeted against PLK1. J. Natl. Cancer Inst., 96(11), 862-72.
Yuan, J., Yan, R., Krämer, A., Eckerdt, F., Kaufmann, M. & Strebhardt, K (2004) Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene 23, 5843-52. 5. Yuan, J., Eckerdt, F., Bereiter-Hahn, J., Kurunci-Csacsko, E., Kaufmann, M. & Strebhardt, K (2002a) Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene 21, 8282-92. 6. Spänkuch-Schmitt, B., Bereiter-Hahn, J., Kaufmann, M. & Strebhardt, K (2002b) RNA silencing of polo-like kinase expression induces apoptosis and abrogates spindle formation in cancer cells. J. Natl. Cancer Inst. 94, 1863-77. 7. Yuan, J., Krämer, A., Eckerdt, F., Kaufmann, M. & Strebhardt, K (2002b) Efficient internalization of the polo-box of Plk1 fused to an Antennapedia peptide results in the inhibition of cancer cell proliferation. Cancer Res. 62, 4186-90. 8. Spänkuch-Schmitt, B., Wolf, G., Solbach, C., Loibl, S., Knecht, R., Stegmüller, M., von Minckwitz, G., Kaufmann, M & Strebhardt, K (2002a) Downregulation of human polo-like kinase by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene 21, 3162-71. 9. Yuan, J, Hörlin, A., Stutte, H.J., Rübsamen-Waigmann, H. & Strebhardt, K (1997) Polo-like kinase, a novel marker for cellular proliferation. Am. J. Path. 150, 1165-1172. 10. Holtrich, U., Wolf, G., Bräuninger, A., Karn, T., Böhme, B., Rübsamen- Waigmann, H. & Strebhardt, K (1994) Induction and down-regulation of PLK, a human serine/threonine kinase, expressed in proliferating cells and tumors. Proc. Natl. Acad. Sci. USA 91, 1736-1740.