

Introduction
The inappropriate repression of genes required for cell differentiation has been linked to several forms of cancer and in particular to acute leukemia. In various acute leukemias, chromosomal translocations lead to the formation of fusion proteins which no longer function as transcriptional activators, but instead repress target genes by recruiting histone deacetylases (HDAC). Similar events leading to aberrant transcriptional repression of target genes are likely to play an important role in solid tumors (e.g. colon carcinoma) as well and corresponding changes of expression patterns should be detectable by gene expression profiling. The modulation of signaling events by histone deacetylase inhibitors has been shown to lead to the induction of apoptosis or differentiation of carcinoma cells. Thus, HDAC inhibitors are candidate drugs for the treatment of cancer (1). The benefit of these inhibitors has been established by in vitro experiments, animal models and in experimental therapy. Nevertheless, the molecular mechanisms underlying these processes are still under intense investigation. Recently, we discovered that the well-tolerated antiepileptic drug valproic acid (VPA) is an HDAC inhibitor (2). This activity can be separated from the antiepileptic activity. Since VPA has been used clinically for more than two decades in the therapy of epilepsy, in-depth knowledge on pharmacology and side effects is available. Our data show that HDAC-dependent transcriptional repression is relieved by treatment with VPA. VPA induces hyperacetylation of histones and inhibits HDAC activity. In the course of an NGFN 1 Cancernet project we found significant proteasomal degradation of HDAC2 protein lin cells treated with VPA but not with other HDAC inhibitors such as trichostatin A. As a consequence, VPA acts selectively on one specific HDAC isoenzyme out of 11 HDACs identified to date (3). Furthermore, we confirmed that HDAC2 can be used as a marker for biological activity in patients treated with VPA.
Currently, VPA is in a clinical phase II trial as monotherapy for advanced malignant diseases at the Krankenhaus Nordwest in Frankfurt (clinical investigator: E. Jäger) and in phase II and phase III trials as combination therapy for acute myeloid leukemia (German AML study group). Biomonitoring of peripheral blood cells under therapy has shown a strong induction of histone hyperacetylation and regulation of known target genes.
Results
For the monitoring of clinical trials it is critical to use a set of reliable biomarkers. In addition to histone hyperacetylation and HDAC2 degradation we identified additional biomarkers including p21, p27, Ubc8, Stat1, TRADD, RLIM, Stat5, Bcl-XL and Survivin. While most of these markers exhibit significant response at the mRNA level, some can only be monitored at the protein level (e.g. HDAC2). We found that histone acetylation is not a reliable biomarker due to large fluctuations in baseline levels.
Our functional studies on the molecular mechanisms of HDAC inhibitor therapy revealed that Stat1 and NF-#B, two key regulators of signal transduction, gene expression and apoptosis are linked via acetylation of Stat1 lysine residues. When we started to study the effects of HDACi on melanoma cell lines, we realized that they can be divided into sensitive and resistant sub-classes. This allowed us to investigate the underlying molecular mechanisms in a set of cell lines derived from the same type of tumor. Our data indicate that sensitive cell lines undergo programmed cell death via both, the extrinsic and the intrinsic apoptotic pathways. In sensitive cell lines HDACi treatment significantly decreases the expression of anti-apoptotic genes such as Bcl-XL, Stat5 and survivin which are bona fide target genes of NF-#B. In resistant cell lines, on the other hand, neither changes in expression levels of these genes nor apoptosis induction are detectable, although hyperacetylation of histones is readily apparent.
A microarray analysis revealed that Stat1 is among those genes which are significantly up-regulated in sensitive melanoma cell lines in response to the HDACi VPA and TSA. These findings were confirmed at the protein level. Interestingly, Stat1 expression was very low and not inducible in HDACi-resistant cell lines. Furthermore these cell lines, in contrast to HDACi-sensitive cells, were not affected by interferon ##. In order to investigate whether Stat1 plays indeed a causative role in the induction of apoptosis in response to HDACi, we introduced Stat1# into resistant cells by lentiviral transduction. Our results show that expression of Stat1 in this cell line restored sensitivity towards HDACi and interferon #. Further experiments are required to establish whether this Stat1-dependent mechanism determining resistance or sensitivity towards HDACi represents a general principle relevant to many types of tumor cells. The HDACi-resistant cell lines were originally established from patients who had undergone immunotherapy including interferon # treatment. It is likely that during this process interferon # resistant cells with defects in Stat1 signaling were selected. In principle both, mutations within the Stat1 gene as well as epigenetic silencing could shut down Stat1 expression. Our observation that resistant cell lines re-express Stat1 when treated with 5-aza-cytidine highlights the relevance of DNA methylation in this context. HDACi and interferon ## are being considered as candidate drugs for cancer therapy. According to our data, the combination of HDACi and interferon # or demethylating agents could be particularly effective in the treatment of melanomas. If this would turn out to be the case, Stat1 expression might serve as a useful marker for the prediction of clinical response.
Stat1 - NF-#B cross-talk
We observed that the expression of a subset of NF-#B target genes after treatment with HDACi or interferon # inversely correlates with Stat1 levels. This prompted us to analyze the affinity of NF-#B for DNA in cells with different Stat1 expression status. A reduction of NF-#B DNA binding was only observed with HDACi-treated or interferon #-stimulated Stat1-positive but not with Stat1-negative cell extracts. This is consistent with the cell-type specific repression of NF-#B-dependent genes by this cytokine and HDACi. Moreover, in cells expressing Stat1, the amount of p65 in the nucleus drops significantly in response to such treatment. Since these results could be due to a physical interaction of Stat1 and p65, we performed co-IP experiments. Indeed, we detected the formation of a Stat1 - NF-#B complex upon treatment with HDACi and interferon #. Gel filtration experiments indicate that the molecular weight of this complex is in the mega-Dalton range. Therefore, the complex is likely to contain several additional proteins. Although a potential cross-talk of Stat1 and NF-#B signaling pathways has been discussed in several reports, evidence for the physical association of these factors has not been published. This is probably due to the fact that a robust interaction can only be observed upon treatment of cells with HDACi or extended stimulation with interferon #.
Acetylated Stat1 mediates suppression of anti-apoptotic NF-#B target genes
Acetylation is considered as a covalent modification which could, similar to phosphorylation affect the activity of a wide range of proteins by altering intermolecular interactions. Transcription factors such as NF-#B and p53 were shown to be acetylated in their DNA-binding domains. We found that acetylation of Stat1 lysine residues 410 and 413 within its DBD regulates its interaction with NF-#B p65. This interferes with NF-#B function and cells are rendered permissive to apoptosis induction (Fig. 1).
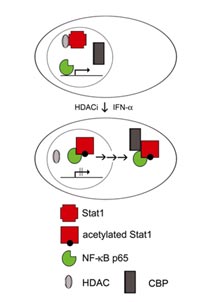
Our data indicate that acetylation of Stat1 and inhibition of NF-#B is required, though not sufficient for induction of apoptosis. Such an observation is consistent with data describing increased susceptibility to apoptosis upon introduction of dominant-negative I-#Bs, yet no apoptosis induction solely by expression of these proteins in solid tumors. Additional actions of HDACi such as the modulation of other signaling pathways and altered cell cycle regulation appear to be necessary. Consistent with this hypothesis, low levels of acetylated Stat1 are detectable in untreated cells but do not lead to spontaneous apoptosis.
Interestingly, microarray experiments revealed that in cells exposed to HDACi about one third of significant changes in gene expression consist of repression rather than activation events. This initially unexpected observation could be due to the induction of transcriptional repressors that do not require HDAC activity to function. Such an indirect mechanism would require de novo protein synthesis. On the other hand, the modulation of cross-talk between Stat1 and NF-#B signaling pathways we discovered is independent of protein synthesis and provides a plausible explanation for the suppression of NF-#B target genes by both, interferons and the inhibition of HDAC activity. The resulting down-regulation of anti-apoptotic genes could be a prototypical example for the HDACi- and cytokine-mediated inhibition of gene expression.
Outlook
For the monitoring of clinical trials we identified several biomarkers Furthermore, our functional studies show how acetyl-lysine moieties may contribute to the temporal and spatial regulation of protein function. An interesting future challenge will be to understand, which other signaling networks rely on protein acetylation events and how they generate in vivo responses to external and internal signals. Alterations of the crosstalk between diverse signaling pathways are likely to play an important role in the response of tumor cells to HDAC inhibitor therapy.
Lit.: 1. Krämer OH, Göttlicher M, and Heinzel T (2001) Histone deacetylase as a therapeutic target. Trends in Endocrinology and Metabolism 12, 294-300. 2. Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, LoCoco F, Nervi C, Pelicci PG, and Heinzel T (2001) Valproic acid defines a novel class of histone deacetylase inhibitors inducing differentiation of transformed and tumorigenic cells. EMBO J. 20, 6969-6978. 3. Krämer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, Brill B, Groner B, Bach I, Heinzel T, and Göttlicher M (2003) The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 22, 3411-3420.