

Introduction
APP is a member of an evolutionary conserved gene family, and homologous proteins have been cloned from C. elegans and Drosophila. In humans and mice, two closely related gene products, termed APP-like proteins 1 and 2 (APLP1 and APLP2), have been isolated. Whereas APP and APLP2 are ubiquitously expressed, APLP1 is restricted to the nervous system. APP undergoes proteolytic processing leading to the secretion of large soluble ectodomains, termed sAPP-alpha and sAPP-beta respectively. Subsequent intramembranous processing of the membrane-tethered APP C-terminal fragments by gamma-secretase cleavage results in the production of Aß and C-terminal stubs. Like APP, both APLPs seem to be similarly processed although the Aß region is not present in the APLPs . Little is known so far about the specific functions of these proteins. The proteolytic processing of APP/APLPs is remarkably similar to that of Notch1, a transmembrane protein that is important in early development. Binding of extracellular ligands to Notch1 stimulates proteolysis and release of its intracellular domain which translocates into the nucleus and regulates gene expression. Thus it is reasonable to hypothesize that APP is involved in a Notch-like signaling pathway. Indeed studies indicate that the cytoplasmic domain of APP (AICD) and of both APLPs, are released, enter the nucleus, and together with Fe65 adapter protein activate transcription [1]. So far, the physiological and/or pathological relevance of this signaling cascade for Alzheimers disease is largely unknown. Neurodegenerative processes have been mainly attributed to the neurotoxic properties of aggregated Aß42, however altered APP processing by gamma-Secretase during the course of AD will also modulate AICD-levels which may thus contribute to the pathogenesis and clinical symptoms of AD. Thus, it will be of primary clinical relevance to identify target genes that are directly or indirectly affected by APP-mediated pathways.
Project Status
Knockout Mice deficient in APP family members
To determine the physiological role of APP and to address the question of whether a loss of APP function contributes to AD, we disrupted the APP gene by gene targeting. APP knockout mice show no spontaneous neurodegeneration, indicating that the loss of normal APP function is not the primary cause of AD. Mutant mice exhibited retarded somatic growth, reduced grip strength, agenesis of the corpus callosum and reduced brain weight, increased sensitivity to kainate-induced seizures and behavioral deficits [2,3] . This mild phenotype was rather surprising in view of the widespread expression and evolutionary conservation of APP, and may reflect functional compensation by related proteins.
To examine this possibility, we generated mice lacking individual or all possible combinations of APP-family members to assess potential functional redundancies within the gene family [4,5] Mice deficient for the nervous system-specific APLP1 protein showed a postnatal growth deficit as the only obvious abnormality and APLP2 knockout mice were apparently normal.
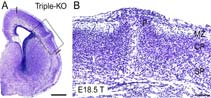
In contrast to these mild phenotypes of single knockout mice, double-mutants carrying APLP2-/-APLP1-/- and APLP2-/-APP-/- -deficiencies proved lethal early postnatally [4]. Surprisingly, APLP1-/-APP-/--mice were viable, apparently normal and showed no compensatory upregulation of APLP2 expression. These data indicate redundancy between APLP2 and both other family members and corroborate a key physiological role for APLP2. None of the lethal double mutants, however, displayed obvious histopathological abnormalities in the brain or any other organ examined. To unravel the full range of physiological functions exerted by this highly redundant gene family, we recently generated mice lacking all three APP-family members to eliminate any residual functional complementation [5]. Triple-mutant mice survive through embryonic development and die shortly after birth. Whereas lethal double mutants showed no morphological abnormalities, we observed a high incidence of cortical dysplasias in triple mutant mice. The cortical dysplasias were characterized by the presence of focal ectopic neuroblasts that had migrated through the basal lamina and pial membrane, a phenotype that resembles human type II lissencephaly. Thus, APP/APLPs play a critical role in adhesion and/or migration of cortical neurons. Moreover, triple mutants showed a reduction in cortical Cajal Retzius (CR) cell number, suggesting that APP/APLPs play a crucial role in the survival of CR cells and neuronal adhesion. Collectively, our data reveal an essential role for APP-family members in normal brain development and early postnatal survival.
Physiological and pathological role of AICD-dependent gene expression
Interestingly a very similar neuronal heterotopia phenotype has been observed in PS-1 deficient mice and double knockout mice lacking Fe65 and the related protein Fe65L1. We thus hypothesize that in wt animals, the APP/APLP intracellular domains may form transcriptionally active complexes that together with Fe65/Tip60 could serve as effector molecule(s) involved in neuronal positioning, possibly via the induction of downstream as yet unidentified target genes.
Further evidence for the physiological role of AICD as an endogenous transcriptional regulator comes from recent work conducted in collaboration with Pardossi-Picard et al. in which we demonstrate that APP-ICD/APLP-ICD containing complexes directly regulate at the transcriptional level the expression of neprilysin a key Aß-degrading enzyme [6]. We show that in APP/APLP2 double deficient cells neprilysin expression and activity is drastically reduced, whereas in familial forms of AD with PS1 mutations neprilysin activity was upregulated. Thus, alterations in AICD-levels, mediated by varying levels of #-secretase activity are a physiological means to modulate Aß-levels and are thus intimately linked to AD pathogenesis.
APP-dependent gene expression
The aim of this study is to identify genes that are affected by Alzheimer-related aberrant processing of APP. In particular we intend to identify further target genes of the signaling pathway that is mediated by the proteolytical release of the intracellular domain of APP (AICD). To this end we employ Affimetrix DNA microarray technology in order to identify genes that are differentially expressed in cells lacking endogenous APP-family members versus samples reconstituted with huAPP variants (huAPP wild type and huAPP-FAD). Since our genetic studies in mice indicated that APP family proteins are highly redundant simple APP overexpression approaches are expected to be confounded by compensatory mechanisms. Therefore, we are using cell/tissue systems that are deficient for endogenous APP/APLPs and reconstitute APP-dependent signaling by expression of huAPP variants. So far, we have established immortalized fibroblast lines derived from double knockout mice lacking both APP and APLP2. These cell lines are functionally equivalent to a triple knockout, as APLP1 is not expressed in fibroblasts. Firstly, expression profiles of APP-/-APLP2-/- fibroblasts are compared to expression profiles of cells stably transfected with huAPP-wt. To this end several individual reconstituted clones expressing huAPP-wt at endogenous level have been obtained. This way we expect to identify the target genes of nuclear AICD signaling and genes indirectly affected by the presence or absence of APP. Analogously, we generated stable transfectants expressing mutant huAPP-FAD (Swedish and London) to compare the respective expression profiles to parental APP-/-APLP2-/- cells, or cell lines expressing huAPP-wt, respectively. We intend to study two sets of huAPP-FAD transfectants: cells expressing huAPP-FAD at the endogenous level (closely resembling the physiological situation in familial AD) and cells overexpressing huAPP-FAD (mimicking the situation of enhanced APP-processing by beta/gamma-secretases in general AD or in transgenic mice, respectively).
Outlook
At a later stage, we also intend to study huAPP-dependent differential gene expression in neurons. For this purpose we will use primary cortical neurons derived from APP-/-APLP2-/- and APP-/-APLP1-/-APLP2-/- triple knockout mice. These neurons will be transduced by lentiviral verctors expressing either huAPPwt or huAPP-FAD. For follow-up analysis we will concentrate on genes matching the following criteria: i) a high degree of differential expression, ii) genes encoding proteins implicated in neurodegeneration, iii) genes that are expressed in the nervous system, iv) genes encoding proteins involved in signal transduction pathways. Differential expression of these genes will be confirmed by quantitative PCR, Northern blotting and in situ hybridization. The complete set of genes will be made available for expression and linkage analysis in AD-patients. This way we expect to identify genes that are affected by aberrant APP-processing during the course of AD, which are expected to play a prominent role in AD pathology.
Lit.: 1. Cao, X. and Südhof, T.C. A transcriptively active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001; 293: 115-20. 2. Müller et al. Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 1994; 79: 755-65.
3. Li et al. Generation of mice with a 200-kb amyloid precursor protein (APP) gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. PNAS. 1996; 93: 6158-6162. 4. Heber, et al. Mice with combined gene knockouts reveal essential and partially redundant functions of Amyloid precursor protein family members. J. Neurosci. 2000; 20: 7951-7963. 5. Herms et al. Cortical Dysplasia Resembling Human Type 2 Lissencephaly in Mice Lacking all Three APP-Family Members. The EMBO J. 2004; 23: 4106 - 4115. 6. Pardossi-Piquard et al. Presenilin-dependent transcriptional control of the Ab degrading enzyme neprilysin by intracellular domains of bAPP and APLP. Neuron, 2005; 46: 541-554.