


Introduction
In current genome research projects, a high percentage of the efforts went into the conception, preparation, implementation and interpretation of gene expression experiments for various diseases. Many groups used cDNA chips from Affymetrix, others used cDNA or DNA arrays spotted with different technologies. These efforts produced large quantities of gene expression data. In each such experiment the chip data, after some statistical normalization, was used to find a relatively small number of genes (e.g. 10 – 500 from the roughly 22.000 on an Affymetrix HG-U133A chip), whose expression level changed most during the measurements. Usually a time series of chip measurements was taken (infected vs. uninfected or normal vs. diseased), then the expression vectors were clustered with methods like hierarchical clustering, self-organizing maps, k-means or other clustering techniques. The underlying assumption was and still is that genes which show a similar change of expression to a stimulus over time belong to a group of co-regulated or closely related genes. This assumption is sometimes, but not always, true. Most evaluation algorithms published during the NGFN funding period dealing with gene expression experiments do not consider existing knowledge of gene regulatory networks or use ontological knowledge in the computer based gene expression analysis phase. This knowledge is only applied afterwards, when interpreting the clustering or classification results by the human experts, who designed the experiments.
Fig 1: Systems Biology – Bridging biology and computer science
Project Status
The toolbox that has been developed since the project’s beginning, namely JCell, is already able to identify the dependencies and the structure of small unknown networks from experimental data by determining the kinetic parameters of the regulatory processes. For this purpose, several mathematical models have been implemented for simulating gene regulatory networks (GRNs). These models can be divided into two classes: first, probabilistic models (e.g. Bayesian networks), which are most suitable for modelling stochastic processes like signalling pathways with only a few reacting molecules. And secondly, deterministic models (e.g. S-Systems) that can be used to model networks in which huge number of molecules are present.
To infer the parameters of those models, JCell mainly uses evolutionary algorithms (EAs) or, if available, straight-forward heuristics. In addition, JCell is able to incorporate biological knowledge about the examined process by automatically importing information from local data sources or public databases in the internet like KEGG. Extended integration of biological data together with the development of new high-performance optimization algorithms will be subject to future work.
Pre-processing
To address large genetic networks, pre-processing of the experimental data is a crucial step in analysis. To reduce the size of the data sets, the first step is to filter genes that did not appear to participate in the biological process of interest, either because their expression signal is below a threshold, indicating experimental noise, or there is so little variation over time. Invariant genes are likely not to be involved in the underlying regulations.
To further reduce the dimensionality of the genetic network inference problem, a widely used method is to cluster similar expression profiles. We have developed intelligent clustering techniques that incorporate biological and especially functional information based on Gene Ontology (GO), a structured and computer processible vocabulary to annotate gene products widely used in all public databases. On the one hand, a functional clustering of genes has been developed that constitutes a tool allowing the biologists to functionally classify their candidate genes. On the other hand, we provide combined clustering methods using both, gene expression data and functional annotation. These methods attempt to facilitate and clarify the interpretation of collected experimental data by monitoring co-regulated genes within their biological context, thus revealing new genetic and functional dependencies which can either be used as expert system or as dimensionality reduction method, as described above, within the inference problem.
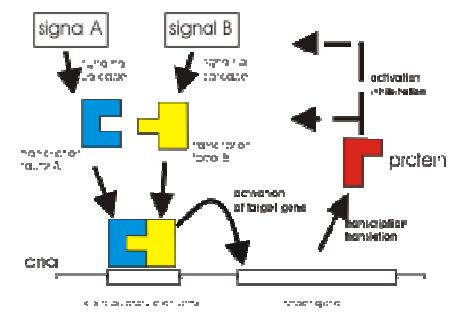
Fig 2: Abstraction of a genetic network used for modeling
They differ in the level of abstraction and the level of complexity. Boolean networks (BNs) are straightforward models that can be efficiently handled with computational methods. But they reduce the gene activity to only two states: either a gene is on (expressed) or off (not expressed), which is biologically not very plausible. However, BNs can be used to examine the mathematical properties of network structures in detail. On the other side of the spectrum, arbitrary differential equations are closer to the true biological process, but they are very hard to handle by means of computer algorithms. Thus, a trade off between abstraction and complexity has to be found.
Currently, several such mathematical models are implemented in the software system JCell:
- Boolean Networks
- Bayesian Networks
- Weight Matrices
- Enhanced Weight matrices
- H-systems
- S-systems
Parameter Optimization
Another major focus of our research are methods to optimize the parameters of the mathematical models. The goal here is to find a set of parameters such that the model shows the same dynamics and dependencies as the examined biological system. Most of the algorithms implemented in JCell are from the field of evolutionary computation, such as genetic algorithms, evolution strategies, or genetic programming, which are well studied optimization method used in many real-world problems. However, we also developed new strategies to optimize the models using special hybrid encoding individuals, graph representations with specialized mutation and recombination operators, and multi-objective optimization that tries to simultaneously satisfy several optimization targets. These extensions are further more using biological data that is automatically imported from public databases like KEGG or TransPATH. With this, networks can be found, which are biologically more plausible than those optimized with standard optimizers.
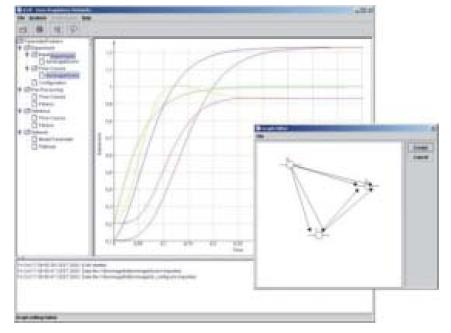
Fig 3: Screenshot of the software framework JCell
The proposed algorithms are currently used for the reconstruction of metabolic networks of E.coli in a collaboration project with INSILICO biotechnology. Further on, we are working on the inference of transduction processes in T-cell signalling and in the modeling of stress-dependent response activity of Arabidopsis thaliana in corporation with researchers
Outlook
Currently, we are working on separation techniques to find sub-systems, or building-blocks like positive or negative feedback loops within the data set rather than trying to model the complete system. Further on, we plan to combine hypothesis-driven methods with data-driven research. As groundwork we build biologically motivated models which are intuitive and capture a high level of detail. The hypothesis-driven modeling effort will form a basis for our data-driven research. Thereby we will integrate heterogeneous experimental observations with biologically motivated models. This task will bring up integration challenges, like multiple testing and meta-analysis issues as well as statistical complexities. We aim to build this modeling framework in a stepwise manner, validating each step from the hypothesis-driven and data-driven perspective, from the very beginning. We think that this approach will not only provide models which explain the regulation of gene expression under various conditions, but also provide models which will be intuitively appealing for scientists with a biological background.
Software
A first version of the software framework has already been implemented and can be freely downloaded from the project’s website http://www.jcell.de. A more recent and extended implementation is available on request.
Lit.: 1. Spieth et al. Predicting Single Genes Related to Immune-Relevant Processes. Computational Intelligence in Bioinformatics and Computational Biology, vol. 2, IEEE press, 2005 2. Spieth et al. Inferring Regulatory Systems with Noisy Pathway Information. LNI, vol. P-71, GI, 2005 3. Spieth et al. Optimizing Topology and Parameters of Gene Regulatory Network Models from Time-Series Experiments, LNCS 3102 (Part I), Springer-Verlag, 2004 4. Speer et al. Functional Distances for Genes Based on GO Feature Maps and their Application to Clustering. Computational Intelligence in Bioinformatics and Computational Biology, vol. 2, IEEE press, 2005 5. Supper et al. Reverse Engineering Non-Linear Gene Regulatory Networks Based on the Bacteriophage # cI Circuit. Computational Intelligence in Bioinformatics and Computational Biology, vol. 2, IEEE press, 2005