

Introduction
The detection of metabolic pathways in classical biochemistry and the more recent observation that many proteins work in form of protein complexes clearly show that proteins exert their function in concert with other proteins. Present research in proteomics, such as genome-wide studies of protein-protein interaction, analysis of the cellular protein topology, and theories on proteome evolution suggests, however, that even pathways and complexes represent only elements of a cell-wide protein network. How this network is organized in the cell, how complex it is, indeed, and what “network” means in terms of the various mechanisms which may relate one protein to another one, are fundamental questions which have to be elucidated. One factor that brings the action of a protein in dependency of other proteins is given by the cellular situation, that all proteins of a cell depend on the same, but limited resources the cell offers its proteins: free space, free water, energy (ATP) and precursor molecules (e.g. amino acids). If a protein is mutated (e.g. knocked out or overexpressed), in case of a disease, this does not only affect the proteins involved in the pathway of that protein (specific effects). In consequence of the disturbed pathway macromolecular crowding may occur in certain compartments of the cell leading to unspecific protein-protein interactions (hydrophobic or electrostatic interactions, surface adsorption). Moreover, protein crowding may decrease the solubility of proteins, leading to conformational changes in proteins with the consequence of self- or heteroassociation, precipitation, formation of plaques or inclusion bodies, as observed in neurodegeneration. Furthermore, local changes in the capacity of energy available for protein activities may affect the rate of metabolic flux in pathways distantly related to the pathway primarily affected (side effects). The final effect that all these processes may have on proteins, are changes in the quantity of proteins. One can assume that the proteome of a cell is quantitatively precisely arranged and well balanced and that the cell functions depend on the relative concentration of each particular protein. We are studying protein levels in the brain of mice which represent models for neurodegenerative diseases (ND); in particular models for Alzheimer's (AD), Parkinson's (PD) and Huntington's (HD) disease. We will also investigate the effect that pharmaca, used against ND, may have on the brain proteins. Our general interest is to detect nodal points in the regulatory network of proteins and rules in the up and down regulation of proteins in cases in which the proteome is affected genetically (by ND) or by environmental factors such as pharmaceutical agents.
Project Status
Methods
To study brain proteins we use a special large-gel two-dimensional electrophoresis technique (2-DE), developed in our laboratory, that allows us to detect up to 10.000 proteins/gel pattern of brain tissues. Mass spectrometry (MS) is used for protein identification. For protein quantification the DIGE-System is used, a new and at present the best method for this purpose. We construct a proteome database that connects all our data under interesting aspects.
Mouse models for neurodegenerative diseases (ND)
We investigate and compare mouse models of three different categories: 1) ND: Parkinson’ disease: Parkin knock out (KO), Pink1 KO, #-syn transgene, MPTP treated mice. Alzheimer’ disease: APP-transgene, APP + PS1 transgene, PS1+2 KO. Huntington’ disease: Htt-transgene. Prion disease: Prion infected mice. Multiple sclerosis: N-CAM KO. 2) Non-ND: Fragile-X syndrome, hyperoxy treated mice, old aged mice. 3) Normal mouse strains: C57BL/6, SPR, 129Vs. So far investigated, proteins changes were found in all cases. More than one hundred alterations were found in prion diseased mice, in old aged mice (100 weeks) and in different mouse strains (protein polymorphisms). In other cases we found between ten to forty protein changes. Most of the altered proteins were identified. By comparison the interesting observation was made that quite a number of protein changes occurred in several diseases (see Table). The altered proteins could be divided in: disease specific, specific for ND, or common ND as well as to non-degenerative neuro-diseases including ageing. Moreover, protein changes as detected in the diseased mice were also found -up to 40%- in certain mouse strains in the form of protein polymorphisms. These results support our network theory: Genetic changes may affect specific pathways, act on nodal points in the proteome network and, moreover, lead to unspecific changes, which may not necessarily have consequences for the disease. In the Parkin-KO mouse and the Huntingtin transgenic mouse the protein change found were investigated in more detail to elucidate pathomechanisms. In the Parkinson mouse 13 proteins were down regulated (only one up regulated), four of these are involved in oxidative phosphorylation in mitochondria. In consequence of the down regulation of these proteins oxidative phosphorylation is reduced and, therefore, the level of reactive oxidative products increases in the cells and affect other proteins, in particular parkin.
Characterisation of the cellular proteome
To study the composition and quantitative arrangement of the cellular proteome, we perform several investigations. (1) We try to identify as many proteins as possible in different regions and fractions of the brain. We found that different brain tissues and cell fractions are more specific with respect to the relative concentration of single proteins than with respect to the protein composition. (2) We identify at present a first set of 500 proteins (spots of our 2-DE patterns) from brain, liver and heart. Preliminary results revealed that only a few percent of these proteins are tissue specific. (3) A basic problem is the relationship between protein number and cell size. Does the cell size increase in early embryonic development, when thousands of proteins become newly expressed? We found that in the course of development the cell size does not increase and the protein concentration is always the same. This could be explained by the observation that the occurrence of new proteins is accompanied by the decrease of proteins already present.
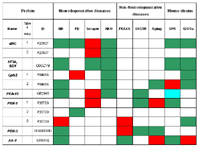
Abbreviations: Proteins: AA-1, Aminoacylase 1; #BC, #B-crystallin; PEA-15, Phosphoprotein enriched in astrocytes 15; PDX, Peroxiredoxin; OXSTR, oxidative stress; SF3b SU4, Splicing factor 3b, subunit 4. Diseases: Aging, 100 week old mice; FRAXA, mice with experimentally induced fragile X syndrome; HD, Huntington’s disease mice; NKO, N-CAM KO mice; OXSTR, mice exposed to hyperoxia; PD, Parkinson’s disease mice; Scrapie, mice infected with prion protein. Further explanations: Mouse strains: Protein polymorphisms are indicated which were found between the mouse strains 129/Sv versus C57Bl/6 and between Mus spretus and C57Bl/6. Red: up-regulation of protein expression, green: down-regulation of protein expression; blue: altered electrophoretic mobility. Comment: #BC and SF3b SU4 were abnormally expressed in mice with neurodegenerative diseases, but not under non- neurodegenerative conditions. When considering, i.e., the Huntington’s disease mouse, out of seven altered proteins found, six were also altered in the SPR-mouse (compared to C57Bl/6), four of them in the same direction (down-regulation).
Outlook
Genetic diseases induced by gene nucleotide mutations, gene knock out or gene transfer begin at the time when the
gene affected would be normally expressed the first time. This beginning is during the embryonic development or shortly after birth. We will study our mouse models in the early stages rather than in adult animals, which express already the disease phenotype. Whereas the proteome in the disease stages only reflect secondary effects due to cell dysfunction, cell degeneration and apoptosis, the early stages may show how the primary changes induced in the proteome are compensated in the development phase, in that the point-of-no-return is still not reached. For elucidating the real origin of a disease and also for therapeutic intervention on the basic level of the disease - not only on the clinical symptoms - this phase is of particular interest. As another aspect, we are interested in studying the relationship between disease-caused protein alternation and protein changes which result from protein polymorphisms. Proteins which show alternations under both conditions may be of particular interest in detecting proteins which modify or impair the course of a disease, or which defend from a disease.
Technical aspects:
To analyse the proteome of pure cell types of the brain (e.g. glia cells, neurons) we at present establish in collaboration with the project group 04 microdissection in combination with protein saturation labeling. Furthermore, we try to simplify our Large-Gel-2-DE for the purpose of commercialisation.
Lit.: 1. Klose J et al. Genetic analysis of the mouse brain proteome, Nat Genet. 2002 Apr;30(4):385-93. 2. Enard W et al. Intra- and interspecific variation in primate gene expression patterns, Science. 2002 Apr 12;296(5566):340-3. 3. Zabel C et al. Alterations in the mouse and human proteome caused by Huntington’s disease. Mol Cell Proteomics. 2002 May;1(5):366-75. 4. Palacino JJ et al. Mitochondrial dysfunction and oxidative damage in Parkin-deficient mice, J Biol Chem. 2004 Apr 30;279(18):18614-22.