

Introduction
Biomolecular Interaction Analysis (BIA) quantifies binding events between proteins (e.g. receptors, enzymes, antibodies), peptides, nucleotides, carbohydrates and small molecules, thus supporting functional genomic approaches. Based on findings provided by genomic analyses, BIA assigns functions to arbitrary listings of gene products within one proteome. In the subproject, “Functional analysis of protein networks”, we try to identify, kinetically characterize and functionally describe proteins in interaction networks within the brain proteome. To this end, established and novel BIA-methods as well as classical methods in biochemistry, molecular biology and cellular biology are used.
Based on the analysis of selected protein networks, a more complete understanding of brain function will be generated and will be implemented into a bioinformatics platform. A focus is the functional characterization of protein-protein interactions in diseased tissues and cells. The relevance of cofactors and posttranslational modifications for protein function is analyzed. Phosphorylation patterns and the kinases involved herein are investigated employing Biacore technology and mass spectrometry. Model proteins, e.g. cAMP-dependent protein kinase and tau-protein are used as model systems for kinases and phosphorylation patterns, respectively.
In close collaboration with partners within the NGFN2 SMP Proteomics and KGs, interaction analysis is performed on an advanced level using state of the art BIA-equipment (Biacore instruments, AlphaScreen, BRET, FRET). Bioinformatics and toponomics will be used to identify relevant interaction partners which are validated in further analyses in vitro and in vivo.
Project Status
Novel BIA-methods have been validated using the cAMP dependent protein kinase (PKA) as model system. The modular structure of this kinase which consists of two regulatory subunits that each bind a catalytic subunit makes it an ideal tool to study protein-protein interactions (1). Since the interaction between the regulatory and catalytic chains is strongly modulated by the second messenger cyclic AMP, the system is also very well suited to analyze the effects of small ligand molecules on macromolecular interactions. This is of special importance in the drug development process, where a specific interaction is typically chosen as a “target” for pharmaceutical compounds (2).
BIA technologies
Surface plasmon resonance (SPR) is still considered the gold standard for high-resolution measurements of any biomolecular interation, i.e. protein-protein, protein-DNA, ligand-receptor or antibody-antigen interaction. The group around Prof. Herberg has a long-standing experience in the application of SPR using the Biacore technology since 1992. In the beginning of 2001, commercialization of the technology was achieved with the startup company Biaffin GmbH & Co KG. Besides the generation of highly reproducible equilibrium binding constants or EC50 values, Biacore allows the determination of separate association and dissociation rate constants for the distinct desription of biomolecular interaction patterns. Typical questions adressed by this technology are “how strong is the binding?”, “how stable is a complex?”, “how efficiently does a competitor challenge a preformed complex?”
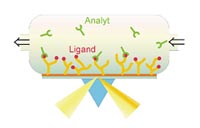
Biacore technology has also been implemented in several platform technologies in proteomics, for example in combination with mass spectrometry (BIA-MS).
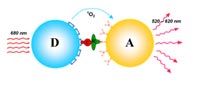
For certain applications, the low throughput inherent to the Biacore technology constitutes a limiting factor. Therefore, alternative technologies have been evaluated. AlphaScreen (Amplified Luminescence Proximity Homogeneous Assay) is a high-throughput capable bead-based assay platform for the detection of biomolecular interactions and permits a rapid screening of pharmaceutical compounds for their efficacy. The detection of an interaction is based on the proximity of donor and acceptor beads (Ø 250 nm), which results in a luminescence signal. If the interaction is disrupted, for example by a pharmaceutical substance, the luminesence signal decreases. This allows us to obtain EC50 values for such compounds, but it lacks the capability of determining accurate kinetic constants (3). The homogeneous assay displays a high robustness with a good signal to noise ratio and can be miniaturized down to the 1536 well format.
Fluorescence polarization (FP), another high throughput method, requires fluorescent labeling of one interaction partner and is used for rapid determination of equilibrium binding constants. Isothermal titration calorimetry (ITC) is a truly label free method which is used to gain additional information about the thermodynamics of a given interaction. Recently, Bioluminescence Resonance Energy Transfer (BRET), a cell based assay, has been implemented to quantify protein-protein interaction in response to low molecular weight (LMW) ligands in living cells (4).
These technologies generate large amounts of interaction data in varying data formats. Therefore the need for a comprehensive bioinformatics analysis became apparent. In close collaboration with Microdiscovery GmbH, a partner in the SMP Proteomics, methods for the evaluation, mining and presentation of binding data are currently in development. Mathematical models designed to overcome a common artifact in BIA, namely mass transfer limitations, have been generated and are being implemented in a sophisticated software package, KinetXpert.
Biomolecular interactions are governed by PTMs
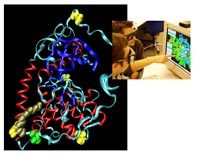
Posttranslational modifications (PTMs) of proteins, like phosphorylation or acylation, exert control over signal transduction pathways in eukaryotic cells. Deregulation of these control mechanisms can lead to severe diseases, among them cancer or neurodegenerative disorders. The most prominent PTM in eukaryotes is phosphorylation of proteins, which is mediated by protein kinases. Very recently, technology has become available to detect phosphorylation patterns with high accuracy. We have established state-of-the-art mass spectrometry (MS) technology to describe the phosphorylation state of proteins qualitatively and quantitatively. Protein kinases themselves are subject to regulation by protein phosphorylation, which is crucial for their correct function. This has to be taken into account, when recombinant expression is used to produce proteins for interaction analysis. Different expression systems have to be tested for their ability to produce the correctly modified protein. Depending on the protein investigated, expression in E. coli may well be sufficient, but more often, eukaryotic expression systems (e.g. Sf9 insect cells, mammalian cells) or in vitro translation systems are required to obtain functional protein. Fig. 4 shows the crystal structure of the catalytic subunit of PKA. Three different types of PTMs are highlighted: Phosphorylation (on four sites), myristoylation and deamidation. Interestingly, for most proteins, neither the stoichiometry nor the stability of the phosphorylation sites is constant over time. This heterogeneity of a given protein species complicates the biochemical analysis and necessitates the detailed characterization of the modification state of a protein under investigation.
Outlook
By means of SPR analysis (and potentially other BIA methods) we aim to detect amyloid peptides etiologic for Alzheimer’s disease in cerebrospinal fluid (CSF) and possibly other body fluids, using specific antibodies. This project is currently done in cooperation with the Friedrich Alexander University, Erlangen-Nuremberg, a partner in the SMP Proteomics.
In close collaboration with the GSF-National Research Center for Environment and Health, Neuherberg, and other partners within the NGFN2, we are in the process of expressing functional domains of a disease relevant protein kinase with the aim of identifying interaction partners and elucidating the mode of action of this protein. Once interaction partners are identified, BIA can be applied to characterize interactions in detail, and possibly to find substances that can influence these interactions.
Furthermore, since the phosphorylation state of proteins is of particular importance for cellular signalling networks, we also aim to optimize protocols for the quantitative analysis of phosphorylation sites using mass spectrometry.
Lit.: 1. Herberg, FW and Zimmermann B. Analysis of protein kinase interactions using biomolecular interaction analysis. In Protein Phosphorylation- A Practical Approach (Hardie, DG, ed.).1999; Vol.2, 335-71, Oxford University Press, Oxford. 2. Zimmermann B et al. Applications of biomolecular interaction analysis in drug development. Targets. 2002;Aug 1;1(2):66-73. 3. Moll D et al. Current developments for the in vitro characterization of protein interactions. in: Proteomics in Drug Research (Marcus, K., Stühler, K., Warscheid, Hamacher, M., van Hall, A. & Meyer, H. E., eds.).2005, in press; Wiley-VCH, Weinheim. 4. Prinz A. et al. Monitoring cAMP dependent protein kinase subunit interaction in intact cells using bioluminescence resonance energy transfer (BRET). FASEB J. 2004; May 14;18(8), C159, 69.18.