

Introduction
Most biochemical processes within and between cells are put into effect by the interaction between proteins, or between proteins and their substrates. The proteome of a cell is the result of controlled biosynthesis, and hence largely (but not exclusively) regulated by gene expression. Vice versa, the transcriptome can be regarded as a sensitive read-out of the proteome or the biochemical state of the cell. Thus, transcriptome and proteome feedback to each other in a highly complex and somehow controlled way. The understanding of this functional regulation is generally limited to distinct signalling or metabolic pathways. To further analyse the mutual regulatory interactions between transcriptome and proteome the simultaneous measurement of expression levels at the RNA and protein levels will be required. In the Human Brain Proteome Project 2 (HBPP2) we will specifically focus on the analysis of progressive changes at the RNA and protein levels in specific brain regions in mouse models for human neurodegenerative diseases.
Project Status
Towards a comparative transcriptomics and proteomics approach
The general feasibilty of such comparative transcriptome/proteome approaches was established in the first phase of the NGFN. We used DNA-chip based RNA expression profiling and 2D-gelelectrophoresis combined with subsequent peptide mass fingerprinting to explore optimal conditions and the general feasibility of such comprehensive gene expression analyses. As a proof of principle we made an assessment of RNA and protein profiles of adult liver and kidney in the mouse. In a follow up project we then used this approach to analyse genetically modified mice lacking a functional delta-like1 (Dll1, Delta1) gene:
1. Analysis of transcriptome and proteome in male mouse liver and kidney: The substantial differences between these two tissues, provided us with a large set of differentially expressed proteins and genes (1). We used this set of differential expression profiles as a tool to address three major questions: (i) Does protein expression correlate with transcriptional regulation for the most differential proteins, (ii) do transcriptomics and proteomics approaches detect functional categories with different preferences, and (iii) does co-regulated gene expression correlate with co-localisation in the genome? Whereas protein analyses mostly identified known metabolic enzymes and structural proteins, transcriptome analysis revealed the differential expression of functionally diverse and not yet described genes. The comparative analysis suggested correlation between transcriptional and translational expression for the majority of
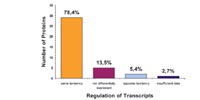
of genes (Fig. 1). Significant exceptions from this correlation confirmed the complementarities of both approaches. Based on RNA expression data from the two hundred most differentially expressed genes, we identified chromosomal co-localization of known as well as not yet described gene clusters (Fig. 2). The high frequency with which we detected such clusters may suggest that co-expression of co-localizing genes is probably rather common.

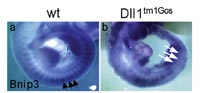
. Analysis of delta-like 1 mutant embryos: The Notch signaling pathway has pleiotropic functions during mammalian embryogenesis. It is required for the patterning and differentiation of the presomitic and somitic paraxial mesoderm and of the neural tube. We used DNA-chip expression profiling and 2D-gel electrophoresis combined with peptide mass fingerprinting to identify genes and proteins differentially regulated in E10.5 Dll1 mutant embryos (2). The differential expression profiling approach identified 47 regulated transcripts and 40 differentially expressed proteins. The majority of these genes has until now not been associated with Notch signaling. Subsequent whole-mount in situ hybridization confirmed that a subset of the identified transcripts has restricted and distinct patterns of expression in E10.5 mouse embryos. For most genes these expression patterns were affected in the presomitic mesoderm, in differentiating somites of Dll1 mutant embryos and in the neural tube and cells differentiating from it (Fig. 3). Similar effects were observed in embryos homozygous for the Headturner (Htu) and pudgy (pu) mutations, which are alleles of the Notch ligands Jag1 and Dll3. The regulated expression of a subset of the proteins was validated by immunoblots. Remarkably six of the proteins down-regulated in Dll1 mutant embryos are proteasome subunits. The large set of regulated genes identified in this differential expression profiling approach is an important resource for further functional studies.
In summary, these initial projects have proven to be useful and will be applied in HBPP2 to study the pathogenesis of Alzheimer´s and Parkinson Diseases.
Mouse models for neurodegenerative diseases
In HBPP2 we mainly focus on mouse models for Alzheimer’s Disease (AD) and Parkinson Disease (PD). Since all currently available mouse models for these diseases do only partially reproduce the phenotypes present in humans, we will analyse several transgenic and k.o. mouse models. As a central model we will concentrate on the Ubb+1 transgenic mice (3), which will be analysed in our consortium for changes in the transcriptome, proteome, toponome, histology, pathology, and other phenotypes. Ubb+1 is a variant of ubiquitin B (Ubb) and is generated from normal Ubb mRNA by a process called ‚molecular misreading’. Enhanced expression of Ubb+1 occurs in human neurodegenerative diseases and can lead to a partial inhibition of the proteasome pathway (4). In transgenic mice, this inhibition causes learning deficits in aged mice, resembling the human situation in AD. In parallel to the analysis by HBPP2 collaborators, the Ubb+1 mice will also be submitted to the German Mouse Clinic (GMC) located at the GSF. The standardized screening methods at the GMC have proven to efficiently and reliably find previously undiscovered phenotypes of mouse mutants (5). We are currently breeding the Ubb+1 transgenic animals under spf conditions in the mouse facility at the GSF and will provide specific brain regions for all HBPP2 collaborators for further analysis.
Expression analysis
Our main interest is the comparative transcriptome and proteome analysis of relevant tissues from mice and humans. As described before, we already have in depth experience with all major aspects relevant for this approach: optimal animal housing conditions, RNA purification and analysis and the generation of proteome data. Technically we provide two independent microarray platforms for mRNA profiling, which can be regarded as largely complementary techniques. Our in house 20k cDNA array is based on the Lion Array Tag Library and has successfully been used to analyse various biological systems (6,7,8). In addition, we routinely use a state of the art GeneChip system from Affymetrix. Both technologies can be applied in HBPP2 to analyse gene regulatory changes, that underlie the development of AD and PD. We are especially interested to analyse early stages of these diseases to identify changes in gene expression, which are causative for disease development. The systematic comparison of our transcriptome analysis with protein expression data will provide us with new information about the complex regulation of biological networks and how gene expression finally translates into phenotypes.
Beside the analysis of relevant mouse models, we are collaborating with the group of Prof. Wiltfang (Erlangen) to detect early markers for AD. Currently, predictive markers for AD can only be obtained from the analysis of peptides in liquor. We set out to find markers in blood samples from patients with mild cognitive impairments (MCI) and markers for AD in their liquor. We will generate transcriptome data from these samples and compare them to data from patients with MCI, but without AD markers. This will be the first time, that expression data from patients with MCI are generated, who’s prognosis for AD has been determined by the analysis of liquor markers.
To gain the maximal benefit from a combined transcriptome and proteome analysis, it will be necessary to tightly link these results to data derived from other techniques provided in HBPP2. To achieve this, we already include further analysis by our collaborators in the experimental design. For example, we are setting up the experimental conditions to profile gene expression from laser microdissected brain regions from histological sections. These sections could then be analysed for their histology and pathology (Prof. Kretzschmar, Dr. Arzberger, GMC) to identify regions with a diseased phenotype. Subsequently, neighbouring sections would be selected to generate data for the transcriptome, proteome (Prof. Meyer, Dr. Marcus), and toponome (Prof. Schubert).
Outlook
Mouse models for human neurodegeneration are indespensable for the molecular analysis of underlying mechanisms. Our combined analysis of the transcriptome and proteome is an efficient tool to link the phenotype of a certain model to the changes in gene expression. This explorative approach will therefore be suitable to find early markers for these diseases and to identify causative genes. The unique combination of a broad range of techniques as present in the HBPP2 consortium will provide us with information from different levels of the biological network in a diseased status. These should allow us to prioritise the differentially expressed genes from our expression analysis and to concentrate on those genes which are prominent in several screens. These genes and proteins, respectivally, can then be validated on the functional level by cell-based assays (Dr. Ueffing) or interaction studies (Prof. Herberg). In addition, we are currently exploring recently developed in silico tools, like pathway analysis programs and the use of standardized Gene Ontology annotations. They will allow us to analyse expression data on a more complex level and will help us to identify affected pathways. This strategy of combining various techniques will result in the generation of information, which can potentially be transferred into clinics.
Lit.: 1. T. Mijalski et al., PNAS 14;102:8621-6 (2005) 2. C. Machka et al., Gene Expr. Patterns Jun 22 (2005). 3. F. van Leeuwen et al., Science 279:242-7 (1998). 4. A.D. Hoppe et al., J. Neurochem. 86(2):394-404 (2003) 5. V. Gailus-Durner et al., Nat. Methods 2, 403 (2005). 6. M. Seltmann et al., Mamm. Genome 16, 1 (2005). 7. J. Beckers et al., Int. J. Cancer 114, 590 (2005). 8. A. D. Greenwood et al., J. Mol. Biol. 349, 487 (2005).