


Introduction
As a relative new and emerging technique, RNAi screening produces a variety of data that poses many unmet informatics needs. From data-handling to standardisation to analysis methods, much remains to be achieved if we are to capitalise on the wealth of information produced. To ameliorate this situation, we established this sub-project with the goal of addressing four of the most pressing issues; 1) automated of RNAi imaging, 2) automated classification of RNAi phenotypes, 3) development of a standardized data model for the phenotype database, and 4) the implementation of a central RNAi phenotype repository.
The automation of the RNAi imaging process presents a number of interesting challenges. To facilitate the smooth automatic capture of RNAi phenotypes, we intend to solve issues such as auto-focussing and object tracking. Real-time classification will also be developed to select positive cells or discard corrupted cells during the image acquisition step. Then, for the automated classification of phenotypes we will model different classifiers for large-scale primary 2D screens and more specialized, secondary 4D screens. Here, our methods will be based on Bayesian learning in combination with artificial Neural Network and Support Vector Machines. We intend to establish an automated workflow for the generation of the phenotype classifiers to distribute the described classifiers to the different SMP partners and clients and to increase the throughput of cellular RNAi assays based on different class stratifications.
All of this work requires considerable efforts on the standardisation side and here we intend to capitalize on existing collaborations with the SMP Bioinformatics to establish quality standards for a unified annotation of RNAi experiments, techniques and phenotypes. These controlled vocabularies will be integrated into the RNAi data model, which will be a further extension of the iCHIP data model developed within the SMP Bioinformatics. Finally, using this data model we will implement an OME compatible NGFN repository of RNAi phenotype data. We also plan to incorporate imaging data from living cell cultures or fixed cell samples into existing applications using the developed standards. The integration of data from microscopic imaging in combination with multidimensional digital analysis will assist functional and comparative studies of proteins in cellular RNAi screens. Automated readout from our applications will be integrated into the database to guarantee comparable classifications of the cellular phenotypic observations.
Project Status
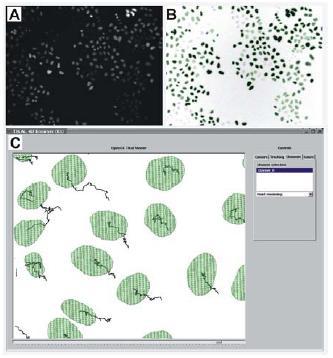
Fig 1: A: original image showing multiple HeLa cell nuclei derived from primary screens of the MitoCheck project. B: result of our segmentation algorithm; the original image (A) is inverted and contrast enhanced; the segmentation result is represented by the green contours; we can see that a high percentage of cells were segmented; only very low contrast cells with grey values near the background noise are missing. C. Tracking result for multi-cell image generated using the TIKAL software.
Significant progress has been made on all fronts since the inception of this project. In collaboration with the Ellenberg lab, we have recently established a fully automated platform for high-throughput cell phenotype screening combining human live cell arrays, screening microscopy and machine-learning based classification methods. Efficiency of this platform was demonstrated by classification of sub-cellular patterns marked by GFP-tagged proteins. Our classification method can be adapted to virtually any microscopic assay based on cell morphology, opening a wide range of applications including large-scale RNAi screening in human cells. The following sections highlight some of the key elements in this work.
Automation of RNAi imaging.
In order to perform the 2D primary screens, we have adapted and extended our existing methods for image feature extraction and classification to the demands of large-scale RNAi screens. As the images that result from high-throughput screens typically contain multiple objects, the segmentation and labelling of single objects of interest is a crucial step for automated analysis. We therefore developed a new 2D segmentation method that segments and labels multi-cell images based on region adaptive thresholding. This technique proved to work fast and reliably and also performed well on images with uneven illumination. In addition we adapted the existing feature extraction methods to process multi-object images. Using the results of the segmentation algorithm, features of single objects can now be extracted directly from the input images. For the classification step we can apply our existing data-mining platform ‘mine-it’. High classification accuracies have been yielded using Support Vector Machines with Radial Basis Function as kernel and cross-validation for optimal parameter search. Thus, at present we can provide an automated workflow to analyze 2D primary screening data that can be adapted to virtually any RNAi screening experiment.
Regarding the more specialized, secondary 4D screens we are currently working on an advanced tracking algorithm that copes with sequences of multi-cell images in 2D and 3D (Fig. 1C). This method will be able to handle splitting cases and return tree-structured tracks in order to analyze the ancestral relationships of mitotic cells. Another current issue is the improvement of the segmentation algorithm described above, with the goal of making it more general and reduce its dependency on user defined input parameters.
Automated classification of RNAi phenotypes.
At present, there are no standardized methods available for an automated phenotyping of RNAi data. Therefore, methods developed in this subproject will be of high interest for researchers on an international scale. To serve current projects on RNAi screening applications through multi-dimensional image data storage and analysis, we will follow the Open Microscopy Enviroment (OME) Standards developed mainly by Jason Swedlow (Department of Gene Regulation and Expression, University of Dundee, UK). Further developments based on RNAi specializations and changes of the data model will be closely synchronized with the Department of Gene Regulation and Expression in Dundee.
For the storage of the image data generated at the GSF at Munich (Prof. Wurst; subproject 7) we will rely on their experience in standards and phenotyping of animal models. Our commercial partner Cenix (subproject 9) has established a fully functioning LIMS system (partly founded through NGFN1) for RNAi data and we will implement this LIMS system for the two RNAi image data production sites (Dresden and Heidelberg). The interface to the local installations of the LIMS system and the RNAi phenotype database includes experimental and technical annotation data transferred from the LIMS system into the RNAi phenotype database. Image data will be only stored once within the central repository and only linked to the local LIMS systems.
The integration of automated image analysis and pattern recognition routines into the screening microscope is carried out in close collaboration with the Ellenberg and Hyman groups, especially for adaptation of movie tracking and registration software modules. The phenotype database was set up based on experience with data management of complex RNAi data collected over the past year in the Hyman lab. In future, the collected RNAi database will be merged with image data gained from RNAi treated animals by Wurst lab. Thus, the utilization of our systems and transfer of technology under strict technology transfer agreements to our industrial partner Cenix is an inherent part of this subproject. One goal of this subproject is to deliver standardized RNAi phenotype data on a genome-wide scale that can be readily delivered for a cross-platform comparison with other genetic repositories and functional databases. Here, we closely interact with the data-analysis task force within the central SMP Bioinformatics.
Standardization of RNAi data annotation and implementation of RNAi database model.
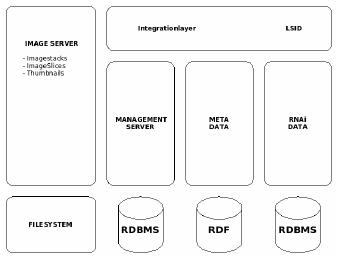
Fig 1: The RNAi database concept.
Besides automatic classification, the correlation of RNAi phenotypes with secondary molecular data and clinical data will play a central role in this subproject. We have currently derived and developed a concept of how to integrate the comprehensive data from genomic wide RNAi screening using the existent OEM standard. Comparable to the OME-System we subdivide the RNAi database into four separate parts (Fig. 2). The first part contains the image data itself. Because of the huge amount of data (typically several of terabytes for a whole genome screen) the raw image data is stored within the file system. The management of this data is carried-out in the second part, the so-called ‘management server’. The description of the images, technical image details as well as RNAi phenotypes will be held in the Meta Data (third part) using primarily controlled vocabularies. These controlled vocabularies will be integrated into the RNAi data model using RDF (Resource Description Framework) technology. This third part will be a further extension of the iCHIP data model developed within the SMP Bioinformatics. Finally, in the fourth and last part, is found the RNAi genomic information. Every database access-point will be available and referenced by the Integration Layer using the LSID (Life Science Identifier). The technology of LSID stands for a unique access approach within Life Science which has mainly been developed by IBM.
Through the standardization of RNAi data, integrated management of RNAi data (especially animal model data) and its correlation with clinical data the present subproject plays an instrumental role in transferring the platform technology into clinical applications. Further, the integrated data-mining workflow enables the clinical user community to correlate complex RNAi phenotypes with clinical data. All data released for public access (according to the publication regulations to be implemented) by the SMP RNAi and by our service clients will be integrated into this central RNAi phenotype repository. This subproject bears the potential to create one of the largest repositories for RNAi phenotypes and will thus contribute significantly to the worldwide standing and visibility of the NGFN.
Outlook
Based on the developed data model we will implement the NGFN repository of RNAi phenotype data. By following our concept of incorporating international standards (see above), the database will be fully compatible with the OME standard. We plan the incorporation of imaging data from living cell cultures or fixed cell samples into existing applications using the developed standards. Automated readout from our applications will be integrated into the database to guarantee comparable classifications of the cellular phenotypic observations. Microscopic images of cell lines as well as of tissue or animal models will be stored in the central RNAi phenotype repository only. An essential excerpt of the LIMS will enrich the central unit to allow inter-experimental studies on genome-wide scale from the RNAi point of view.
Our future work in automatic classification methods will focus on the development of methods concerning the secondary screens. Having established the 3D tracking algorithm, a fast and robust method for 3D segmentation will be required. Furthermore, novel object-related and time-resolved features have to be developed to find discriminative features for RNAi phenotype classification in 3D image sequences. The overall goal will be to establish a software platform that incorporates all tools for the generation of phenotype classifiers to automatically analyze image sequences resulting from large-scale RNAi screens. We intend to distribute the described classifiers to the different SMP partners and clients and to increase the throughput of cellular RNAi assays based on different class stratifications.
In summary then, the overall goal of this subproject is to deliver standardized RNAi phenotype data on a genome-wide scale. This data will be readily delivered for a cross-platform comparison with other genetic repositories and functional databases. Our work within the context of the SMP Bioinformatics will greatly facilitate us in these goals.
Lit.: 1. Watanabe H et al. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature. 2004 May 27;429(6990):382-8. 2. Hampe J et al. Association of NOD2 (CARD 15) genotype with clinical course of Crohn's disease: a cohort study. Lancet. 2002 May 11;359(9318):1661-5. 3. Huehn J et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004 Feb 2;199(3):303-13. 4. Goldberg I, Swedlow JR et al The Open Microscopy Environment (OME) Data Model and XML file: open tools for informatics and quantitative analysis in biological imaging. Genome Biol. 2005;6(5):R47. Epub 2005 May 3.