


Introduction
Changes of DNA methylation play a central role in carcinogenesis. Extensive hypermethylation and consecutive transcriptional silencing of tumorsuppressor genes has been documented in multiple tumor entities. The identification of target genes silenced by this modification has great impact on diagnosis, classification, definition of risk groups and prognosis of cancer patients. Initial genome wide screening approaches by RLGS (restriction landmark genomic scanning) have estimated that approximately 600 de novo methylation events occur within a tumor genome (1). The development of a microarray based technology known as differential methylation hybridization (DMH) (2) has greatly improved the genome wide search for hypermethylated genes and has been successfully used to identify epigenetic signatures of several tumor entities. Based on this protocol we have developed a novel microarray containing 7680 CpG rich fragments of the human genome that can be used to investigate epigenetic alterations in genomic DNA. In the context of the �Epigenetic SMP� it is our goal to continuously expand and improve this microarray technology to cover all cancer relevant CpG islands of the genome.
Project Status
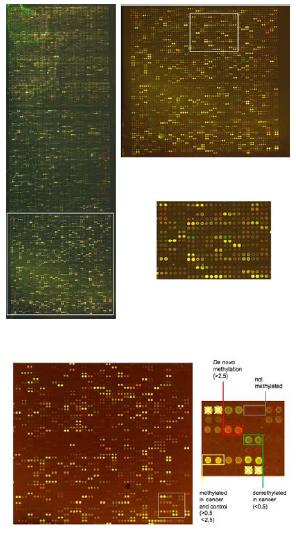
Fig 1: Image of a CpG island microarray containing 7680 individual CpG rich sequences printed as duplicates. The Microarray was hybridized with methylation specific amplicons of a Hodgkin lymphoma cell line (upper panel) and a primary mammary carcinoma (lower panel) and corresponding normal DNA samples.
Applied technology
We have optimized a microarray based screening procedure (DMH, differential methylation hybridization) to detect methylation changes on a genome wide scale. Currently 7680 CpG rich fragments of the human genome were isolated from a human CpG island library and the presence of methylation sensitive restriction sites (BstUI and HpaII) relevant for this technology has been confirmed by restriction digest. In addition most clones with high identity to repeated sequences have been eliminated by colony hybridization to human Cot-1 DNA. PCR products of single copy clones were generated and spotted on UltraGap glass slides (Corning) using a Q-array mini microarraying system (GENETIX). Each spot is localized using a grid that excludes remaining repeat sequences determined by hybridization of the microarrays with human rDNA and human Cot-1 DNA. A protocol for generation of methylation specific genomic probes from tumor DNA and control tissue has been developed and optimized for 2�g of genomic DNA. DNA is digested with the frequent cutting enzyme MseI (TTAA) leaving CpG islands intact. Following adapter ligation the fragments are digested subsequently with the methylation sensitive restriction enzymes BstUI (CGCG) and HpaII (CCGG). Only methylated fragments remain intact and can be amplified using adapter sequences as primers. Amplified MseI fragments of tumor DNA are subsequently labeled by direct incorporation of CY5-dCTP in a random priming reaction using the Kleenow fragment. Control tissue is treated accordingly and labeled by direct incorporation of CY3-dCTP. Cot-1 DNA is added to both amplicons prior to hybridization. The microarrays are scanned with a GenePiX 4000B scanner (Axon) and signals are located and analyzed by the GenePix 5.0 software (Fig 1). Fragments with exclusive methylation in the tumor genome appear as red spots (ratios Cy5/Cy3>2.5 are scored as methylated) and with exclusive methylation in the control tissue as green signals. Single copy sequences methylated in both samples appear as yellow spots. Several repeat sequences are also contained in defined positions on the microarray and appear as bright yellow signals. These spots are used to normalize the microarray. After normalization the microarray data is imported into the Aquity 3.1 software for subsequent statistical analysis. The sequence identity of spots with differential methylation signals can be determined by automated sequencing of the respective genomic fragment from the bacteria clone and comparison to online databases.
Collaborations within the NGFN2 and other groups
The 7K version of the CpG island microarray is currently used for studying differential methylation in several cancer entities (e.g. gliomas, medulloblastomas, breast cancer, lymphomas, leukemias, prostate cancer, colon cancer) and tumor derived cell lines (e.g. gliomas, medulloblastomas, neuroblastomas) to uncover epigenetic alterations during development of chemoresistance or other cancer related properties (3, 4). Identified sequences are then compared to online databases to identify epigenetically silenced target genes. The methylation status of relevant CpG sites is subsequently verified by sequencing of cloned bisulfite PCR products. The genes identified so far code for proteins with various cellular functions incl. transcription factors, regulators of cell proliferation and cell adhesion molecules (Fig. 2). In addition to the human CpG island microarray we have assembled 7680 murine CpG rich fragments used by groups of the NGFN2 network and other scientists. These microarrays are especially useful for in vivo analysis of DNA methylation. The current projects investigate alterations in CpG-methylation during differentiation of stem cells or in mice that are deficient for genes coding members of the epigenetic machinery.
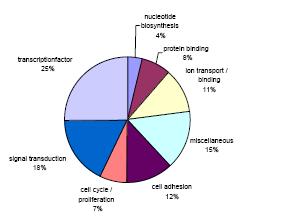
Fig 2: Representation of de novo methylated genes identified in the investigated cancer entities by their cellular function.
Epigenetic alterations have been described as powerful markers for the early detection, classification and prognosis of cancer. There is a major focus within this SMP to use specific CpG sites as diagnostic markers. The identified sequences of our project will be potentially useful for the development of such assays. In particular the identification of methylation profiles in distinct cancer subtypes by the same technology may help to identify common themes of epigenetic silencing as well as elucidate highly specific markers for molecular diagnostic analysis. By this means it will also be possible to discriminate between differentiation specific methylation changes and epigenetic alterations associated with neoplasia. For this we want to assemble a database containing all methylation data acquired for all analyzed cancer entities. This goal will be approached by using the novel technique of differential methylation hybridization (DMH) and related technologies that allow comprehensive analysis of the methylation status at multiple CpG islands distributed along the entire genome.
Outlook
Epigenetic alterations have been described as powerful markers for the early detection, classification and prognosis of cancer. There is a major focus within this SMP to use specific CpG sites as diagnostic markers. The identified sequences of our project will be potentially useful for the development of such assays. In particular the identification of methylation profiles in distinct cancer subtypes by the same technology may help to identify common themes of epigenetic silencing as well as elucidate highly specific markers for molecular diagnostic analysis. By this means it will also be possible to discriminate between differentiation specific methylation changes and epigenetic alterations associated with neoplasia. For this we want to assemble a database containing all methylation data acquired for all analyzed cancer entities. This goal will be approached by using the novel technique of differential methylation hybridization (DMH) and related technologies that allow comprehensive analysis of the methylation status at multiple CpG islands distributed along the entire genome.
Lit.: Costello JF et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000 Feb;24(2):132-8. 2. Huang TH et al. Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet. 1999 Mar;8(3):459-70. 3. Waha A et al. Methylation profiling identifies epigenetic markers for high-grade gliomas. Cancer Genomics and Proteomics. 2004 (1), 209-214. 4. Waha A et al. Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia. 2005 (7), 193-199.