


Introduction
There has been a tremendously increased interest in DNA based diagnostics, since the investigation of molecular biological mechanisms has been extended to the genomic level and after the sequence of the human genome has been established. While most research is currently being devoted to the discovery and clinical application of genetic, proteomic and RNA expression markers, DNA methylation is rapidly emerging as a new level of cellular information. DNA methylation is involved in the regulation of gene activity. Aberrant DNA methylation has been associated with a variety of human diseases, and DNA methylation markers hold a great promise as a diagnostic tool, e.g. in early cancer screening [1]. In contrast to expression markers DNA methylation markers are accessible at the DNA level which enables their analysis even in archived paraffin-embedded tissue samples. Methods are in place to detect a few copies of aberrantly methylated DNA in a background of a vast excess of normally methylated DNA which is the precondition for assaying methylation markers also in body fluids that are readily accessible in clinical practice.
Several methods are available for the genome-wide discovery of methylation markers differentiating between different types of tissue samples [1]. They are mainly based on the use of methylation specific enzymes. The most prominent examples are restriction landmark genomic scanning (RLGS), representational difference analysis (RDA), methylated CpG Island amplification (MCA)-RDA, methylation sensitive arbitrarily primed PCR (MS-AP-PCR) and differential methylation hybridization (DMH) (Ushijima, T; 2005). However, non of these published methods fulfil all of the following important requirements:
- usage of small DNA sample amounts
- viability for analysis of paraffin embedded tissues
- real genome-wide coverage
- viability for parallel analysis of unlimited number of samples
- high reproducibility
- fast and easy handling
Project Status
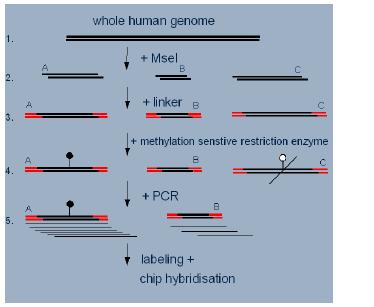
Fig 1: Workflow of the DMH-amplicon preparation.
Together with an optimised amplification and labelling protocol this workflow was successfully applied to the comparative methylation analysis of peripheral blood lymphocytes and a breast cancer cell line (Fig. 2). A custom made Affymetrix microarray carrying an initial selection of features designed for protocol optimisation was used to analyse the amplicon. First results indicate that the optimised preanalytics workflow generates promising marker candidates and can be conducted in a highly reproducible manner.
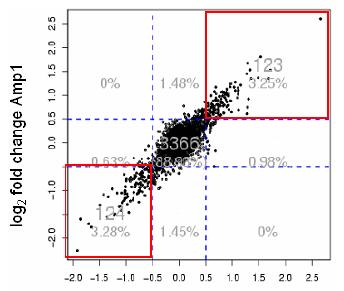
Fig 2: Correlation plot of two DMH discovery experiments using PBL and breast cancer cell line samples. Spots in the red boxes indicate potential marker fragments reproducibly found in both experiments to be either methylated or unmethylated in breast cancer cell line compared to the PBL.
Although a DMH utilizing the optimised preanalytics workflow in conjunction with a second generation custom-made microarray is expected to deliver methylation marker candidates at a low false positive rate the validation of these candidates on hundreds of clinical sample is a vital step of the marker development process. Different techniques are available for analysing DNA methylation at individual loci [1], most of them based on bisulfite conversion of sample DNA. At Epigenomics a methodology for the parallel methylation analysis of hundreds of CpG positions based on the hybridisation of bisulfite PCR products onto arrayed oligonucleotide probes has been developed [3] (Figure 3). This method has been successfully applied to the validation of candidate methylation markers in patient sample sets.
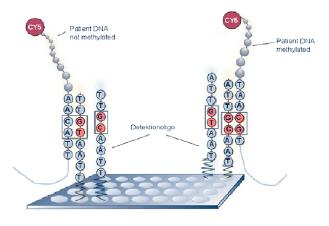
Fig 3: Methylation analysis by bisulfite PCR product hybridization onto arrayed oligonucleotide probes.
In the framework of the NGFN2 project it is planned to evaluate different array platforms based on different principles for signal generation for their potential for methylation analysis via hybridisation of bisulfite PCR products. Once a suitable analytical platform has been identified an integrated workflow combining all steps from sample preanalytics to data analysis will be established and optimized using established reference markers. The ultimate goal is to establish a platform which enables clinical studies based on methylation marker analysis to be performed in a highly standardized manner in a sufficient throughput to support the development of diagnostic tests.
Lit.: Lit.: 1. Laird PW. The Power and the promise of DNA methylation markers. Nature Rev. Cancer 2003;3:253-66. 2. This work was supported by the BMBF Foerderprojekt 0313030C Technologien zur Charakterisierung des humanen regulatorischen Proteom. 3. Adorjan P. et al. Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Research, 2002, 30(5), e21.