


Summary and Major Objectives
Haplotype-based approaches to disease gene discovery have become a central theme. The 'International HapMap Project' has been launched to facilitate discovery of sequence variants that affect common disease and establish new routes to diagnostics and therapeutics of immense medical benefit (Nature 425: 758-9, 2003; Nature 426: 789-96, 2003). This project relies on the assumption that the human genome can be resolved into 'blocks' of common haplotypes, with only few haplotypes per block and few SNPs necessary to tag each block, allowing genome-wide association and candidate gene studies at much higher efficiency. With view of future lines of investigation it has been recognized that the evaluation of high resolution genetic variation data will be the next important and necessary step in order to 1) assist optimisation of SNP selection and analysis of LD and haplotype structures and extraction of tags and 2) systematically assess the 'completeness of the information' (Nature 426: 793, 2003). In depth knowledge on the amount and nature of information that will be added will be indispensable and critically reflect on the power of current haplotype approaches to represent underlying LD and haplotype structures and their validity as a tool to map causative variants. It will, moreover, critically guide the design of the ultimately successful approaches to haplotype-based disease gene discovery. It will, at last, provide the basis to make informed decisions on the meaningful investments in this line of research in the future.
Our major objective is to perform a first systematic investigation in this direction, analysing high-resolution genetic variation data in comparison to the data provided by the HapMap Project. The following prerequisites will provide the necessary basis to carry out such an analysis competitively: a) High resolution data sets obtained by the comparative sequence analysis of nuclear loci in an average of several hundred individuals including cases and controls, an unprecedented depth, up to two orders of magnitude deeper than previous resequencing studies in comparable populations; b) a novel, highly efficient haplotyping technology (CSH), which allows the genome-wide determination of the molecular haplotype structures of any gene or chromosomal region, respectively, and c) an (inter)national network of leading experts in haplotype analysis. We will establish a reference resource of haploid clone pools from a total of 250 individuals (500 haploid genomes) from a representative German population. We will type the same loci, using the high resolution-derived SNPs on the one side, the HapMap-derived SNPs on the other side, in the same sample of haploid genomes. We will then systematically analyze and compare the LD and haplotype structures and tag SNPs derived by these two approaches, respectively. Major objectives are: 1) to evaluate to which extent the HapMap-derived SNPs and tag SNPs in fact capture the LD and haplotype structures given at the ultimate level of resolution, DNA sequence, and, in particular, candidate gene-related haplotype structures; 2) to assess, which types of information will be added at increasing levels of resolution; 3) to test at given data sets whether the disease associations derived by high resolution analyses could have been captured by HapMap-derived SNPs and to which extent evaluation of rare (disease-related) haplotypes may be of relevance. Moreover, proposed haploid reference system provides the basis to systematically assess the correspondence of haplotype structures predicted in silico with their molecular correlates. Thus, it will serve as basis to comparatively evaluate, develop, optimise and validate algorithms, an issue of increasing importance with view of the increasingly complex data sets expected in the future.
This study is carried out in collaboration with a team of leading international and national experts on various aspects of haplotype analysis including Dr. J. Ott (Rockefeller University), Dr. R. Shamir (Tel Aviv University), Dr. G.M. Church (Harvard Medical School), Drs. H. Zhao and K.K. Kidd (Yale University), Dr. K.M. Weiss (Penn State University), Prof. J. Reich and Dr. K. Rohde, MDC (bioinformatics); close interactions exist with the SMP-GEM 'Haplotypes in association studies' (Prof. M. Baur, Prof. T. Wienker), Prof. Dr. H.-E. Wichmann and Prof. Dr. T. Meitinger, GSF (haplotype analysis), Prof. S. Schreiber, Uni Kiel (genotyping), Dr. M. Platzer, Dr. S. Taudien, IMB (candidate gene resequencing, SMP1); Dr. T. Sander, NeuroNet; Prof. A. Ullrich, CancerNet (resequencing).
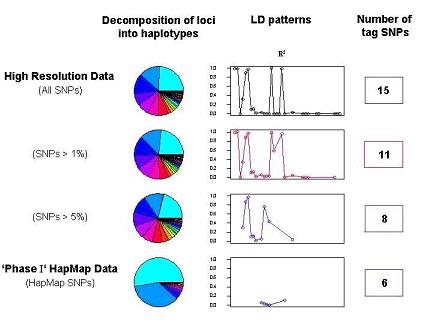 |
| Fig 1: Example of frequency distributions of reconstructed haplotypes, LD patterns (r2) and tag SNPs for a locus using HR SNPs (all HR SNPs, all common SNPs with a minor allele frequency >1% and >5%) and ‘Phase I‘ HapMap SNPs, which represent a subset of the HR data. |
Research Background
A series of studies has shown that human genetic variation has a 'haplotype block structure' such that each chromosome can be decomposed into large blocks with strong LD and relatively few haplotypes, separated by short regions of extensive recombination. Altogether, two to five haplotypes per block, accounting for a total of 75-98% of all chromosomes, were observed, with about 80% of all haplotypes occurring globally. Because SNPs in strong LD carry redundant information, a small subset of common SNPs (‘tag’ SNPs) selected from each segment might suffice to define a limited number of common haplotypes useful in any population. This rationale provided the basis for the development of the 'HapMap', a set of SNPs that capture the haplotype block structure of the genome, so that genome-wide association studies can be carried out most efficiently. In its first round, the 'International HapMap Project' aims to genotype ~ one million SNPs with a minor allele frequency of at least 5%, spaced at approximately ~ 5 kb intervals, in 270 DNA samples including 150 unrelated individuals from four populations (Nature 426: 789-96, 2003). At the end, upon determination of LD and haplotype structures and efficient selection of tag SNPs, genotyping 200,000 up to one million tag SNPs across the genome is expected to capture most of the information about genetic variation and LD represented by the 10 Mio common SNPs (>1%) in the population. Importantly, this approach to haplotype analysis is based on SNP data that have primarily been generated by 'random', genome-wide discovery efforts in limited numbers of individuals (about 20 in average) and spot sampling in several kb-intervals (Hoehe, Pharmacogenomics 4: 547-70, 2003).
Once genetic variation is resolved systematically at the ultimate level of resolution, DNA sequence, however, a substantially different and much more complex picture of given variability, LD and haplotype structures within defined segments of DNA emerges. As demonstrated by a series of studies that have compared candidate gene sequences in higher numbers of individuals, sequence diversity is remarkable, in average ~ one SNP every 170-180 bp. Potentially large numbers of haplotypes are given, up to 88 summarising present results, with an average of ~ 14 per gene in the most comprehensive gene survey. Our own analyses, which reached to our knowledge the greatest depth to date, yielded 16-140 haplotypes per gene. The decomposition of loci into different and less common haplotypes appeared much stronger and the fraction of rare haplotypes was potentially substantial. Haplotypes conferring risk to complex disease obviously were in several instances not part of specific common haplotypes, but part of the fraction of rare haplotypes. LD patterns in any particular genomic region appeared unpredictable, with too little LD over a few kb and too much at greater distances. A fraction of SNPs did not allow inference on flanking sites. Evidence for 'intricate, hierarchical patterns of LD' suggested that association mapping could be problematic, even within a single gene. To summarise, these studies led to the conclusion that an accurate knowledge of patterns of variation and LD between marker alleles as the basis for the selection of representative subsets of sites will be mandatory in order to design meaningful association mapping approaches (Hoehe, Pharmacogenomics 4: 547-70, 2003).
It is now of utmost importance to be able to define the specific relationships between these two different scenarios obtained by different approaches to (and different levels of) resolution. This can obviously most precisely and informatively be achieved by subjecting the same loci to these different approaches. Key questions will be a) the extent to which HapMap-based approaches capture the LD and haplotype structures obtained at high resolution (HR); b) whether HR data just 'add detail', deepening the clades captured by common 'signatures', or c) whether/which qualitatively different information will be added; d) the nature/amount of information added by the systematic analysis of genic SNPs, which represent only a comparably small fraction of SNPs in public databases. These investigations may allow a much more informed evaluation of haplotype-based mapping scenarios in disease.
Project Status
We have performed a first systematic investigation in this direction, analyzing high-resolution genetic variation (HR) data in comparison to the corresponding HapMap-related data (‘Phase I’ HapMap, public data release #16c.1, June 2005). The analyses have been carried out at the example of 28 candidate loci, which have been systematically resequenced in an average of 333 individuals. Specifically, we have comparatively evaluated the numbers and characteristics of both all SNPs and common SNPs, the numbers and relative frequencies of haplotypes, the patterns of LD and the tag SNPs. The first results indicate substantial differences in the major lines of investigation between the ‘Phase 1’ HapMap and HR (figure 1) with significant implications for ‘Phase 1’ map application and future map development (Hoehe et al., 2005, to be published).
Outlook
Undoubtedly, the proposed project will provide essential information for all present and future disease gene discovery projects in the NGFN that rely on genetic variation/haplotype approaches. The results will have important implications on the development of successful strategies and investment of resources. Importantly, this project implies the establishment of a 'community resource', accessible to any collaborators from the national/international genome networks: a resource for the validation of haplotype structures, a reference system for the development of algorithms and a 'permanent' control group and reference resource for all NGFN2 SNP and haplotype-based disease association studies. Moreover, access to the molecular haplotypes of any gene/potential drug target in a population of substantial size will provide essential information to pharmaceutical and biotech companies, which will help elucidate individually different drug response and facilitate processes of drug target evaluation, prioritisation and clinical trials. Thus, it represents a key resource for pharmacogenomic approaches to drug development. Proposed haploid reference resource represents the basis 1) to test for existence of numerous individually different forms of a gene and 2) to provide the templates for their in vitro functional characterisation. This represents a key step in the evaluation of gene function, dysfunction, the molecular basis of drug response and disease processes.
Lit.: 1. Hoehe MR. Haplotypes and the systematic analysis of genetic variation in genes and genomes.Pharmacogenomics. 2003 Sep; 4(5): 547-70. 2. The International HapMap Project Nature. 2003 Dec 18; 426(6968): 789-96. 3. Dennis C.The rough guide to the genome. Nature. 2003 Oct 23; 425 (6960): 758-9. 4. Hoehe MR et al. Sequence variability and candidate gene analysis in complex disease: association of mu opioid receptor gene variation with substance dependence. Hum Mol Genet. 2000 Nov 22; 9(19): 2895. 5. Burgtorf C et al. Clone-based systematic haplotyping (CSH): a procedure for physical haplotyping of whole genomes.Genome Res. 2003 Dec;13(12):2717-24. 6. Branson R, Potoczna N, Kral JG, Lentes KU, Hoehe MR, Horber FF. Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003 Mar 20; 348(12): 1096-103. 7. Hoehe MR et al. Human inter-individual DNA sequence variation in candidate genes, drug targets, the importance of haplotypes and pharmacogenomics. Curr Pharm Biotechnol. 2003 Dec; 4(6): 351-78. Review. 8. Zhang J, Vingron M, Hoehe MR. Haplotype reconstruction for diploid populations.Hum Hered. 2005; 59(3):144-56. 9. Hoehe MR, Kroslak T. Genetic variation and pharmacogenomics: Concepts, facts, and challenges. Dialogues Clin Neurosci. 2004,6