


Introduction
Deciphering the molecular control mechanisms behind the differential regulation of gene expression is a major challenge in functional genomics. Recent research activities have focused on the identification of promoter sequences where DNA signals are thought to determine the specific response to certain environmental and developmental signals. To elucidate these regulatory mechanisms a host of experimental approaches is currently being developed. Bioinformatics supports these efforts in all aspects of experimental design, data storage, integration, processing and analysis.
One problem of particular importance is the precise definition of promoter regions ethods. The explicit goal of this project is to guide the experimental research activities within the NGFN by providing suggestive lists of candidate promoter regions that have varying degree of support from cross-species analysis. Specifically we aim to establish four different categories of promoters
- 1) Approximately 3000 genes with experimentally mapped transcriptional start site in mouse and human.
2) About 8000 genes where the start site was mapped at least in human.
3) A set of orthologous gene pairs with high degree of upstream similarity.
4) The intersect of 2) and 3) which allows promoter prediction in orthologous mouse genes.
All our prediction will be made available to NGFN partners through a web-based interface. We will provide this data for inspection of specific genes, as well as genome-wide downloads suited for large-scale analysis.
Project Status
CORG Database
We have developed a comprehensive database of conserved regions in the human genome and various other vertebrate genomes – the Compartive Regulatory Genomics database (CORG). The main conceptual basis behind this database is that of “phylogenetic footprinting”, according to which one would expect biologically functional sequence elements to be more conserved than the divergent background. For the detection of such conserved regions we have developed sophisticated alignment tools and complemented them with a sound statistical analysis. To fully exploit this data for the purpose of promoter predictions, we started to annotate these conserved regions with other functional sequence information from experimental works (such as experimentally mapped transcriptional start sites) and in-silico predictions (such as conserved transcription factor binding sites).
So far we have included experimentally annotated transcriptional start sites from two other standard databases: the Eukaryotic Promoter Database (EPD) and the Database of Transcriptional Start Sites (DBTSS). Such information may ultimately reveal alternative promoters and transcriptional start sites. Given the recent interest in regulatory element detection away from the proximal promoter, we have also extended our initial study of upstream regions to include conserved sequence elements in the intronic regions and 3’ untranslated regions downstream from the last exon. Finally we suggest conserved transcription factor binding sites based on known motifs from the TRANSFAC database. Since vertebrate sequences often include many redundant repeats we filtered those elements prior to the annotation with transcription factor motifs.
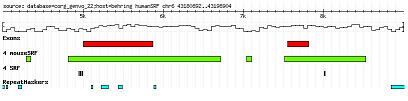 |
| Fig 1: We use an interactive and flexible interface to visualize the putative promoter region of a selected gene and annotate the conserved regions with known transcription factor binding sites. Here an example of human-mouse conserved SRF binding sites is shown in the promoter region of the SRF gene. |
A key issue for the representation and analysis of heterogeneous data from diverse sources is the adoption of certain common standards, which allow for portability and interchangebility of existing annotations. We decided to adopt the controlled vocabulary of the Sequence Ontology for all our sequence features and incorporated this into our annotation pipeline. Moreover, we have decided to represent all our sequence-based annotations according to the GFF3 specification, which is commonly used by other groups.
Modularisation
The steady flux of novel genomic information and sequence annotation from experimental and computational predictions requires a flexible and robust pipeline to incorporate and represent user-specified data in our framework. To this end a number of modules and routines are currently being developed to facilitate the task of importing and exporting data, as well as the annotation steps and data analysis. These factory modules are written in open-source BioPerl and form the core of the BioMinerva management system, which we are constantly developing.
Visualisation
To enhance the communication between bioinformaticians and molecular biologists we are planning to improve the web-based front end of our database, which allows a detailed and customisable visualisation of selected promoter regions. This interface will represent the reference sequence with its annotations such as conservation, transcription factor binding sites, CpG-islands and other selectable features, which may also include external links and references. This infrastructure is based on a Generic Genome Browser (www.gmod.org/ggb) and is currently being developed and tested for a human prototype sequence with a particular focus on promoter-relevant information.
Predictions
1. In collaboration with the Lehrach department at the Max Planck Institute for Molecular Genetics, we have defined a set of putative promoter regions for genes on the human chromosome 21. The predictions for these genes are based on their upstream conservation pattern with respect to mouse orthologous genes and suggest a basis for future experimental validation. We have further annotated these promoters regions with a large set of putative binding sites for transcription factors with known motifs, which suggest molecular mechanism of gene regulation.
2. Conserved non-coding regions lend themselves to annotation with functional elements, such as transcription factor binding sites. In collaboration with the Herrmann department at the MPI-MG we focus on the prediction of transcriptional networks, which are thought to drive important processes in early mouse development and apoptosis.
Using a selection of known transcription factor binding motifs (from literature and the TRANSFAC database), we predicted possible target genes, which are both tailbud-expressed and show an overrepresentation of relevant binding sites in conserved upstream regions or other non-coding regions (such as intronic regions or 3’ UTRs). Many putative target genes show multiple conserved binding sites, suggesting complex combinatorial control mechanisms.
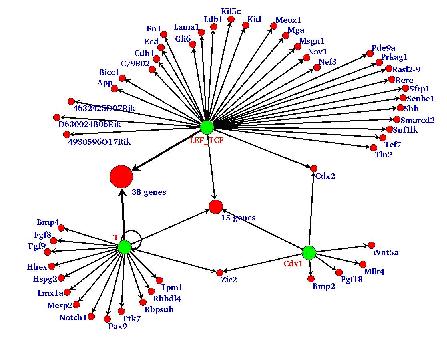 |
| Fig 1: A putative network of transcriptional regulation involving targets of Wnt-signalling in early mouse development. Key transcription factors are represented as green circles, while putative target genes are shown in red. A number of targets (shown as large red circles) suggest combinatorial control through several transcription factors. |
Data Integration
The main task and challenge in the development of our promoter database will be the incorporation of additional promoter-related sequence information, which is to appear as additional tracks in the visualisation. Extending our current annotations of orthologous sequences with conserved non-coding block, we plan to also incorporate whole-genome alignments provided by USCS (http://genome.ucsc.edu/). The latter also considers the synteny of an alignment in its overall chromosomal context. In higher eukaryotes genes are often organised in several possible transcripts, each with their own promoter. To account for this important diversity we also plan to include transcript information from the ENSEMBL database (http://www.ensembl.org). Moreover, we will incorporate transcript information based on our own SpliceNest database (http://splicenest.molgen.mpg.de), which was developed in-house to highlight the tissue-specific character of gene expression based on expressed sequence-tags (EST). We also anticipate novel and large-scale experimental data on transcriptional start sites in mouse from the FANTOM project (http://fantom.gsc.riken.jp), which will be integrated into our promoter database. Computational promoter prediction algorithms often utilize information on CpG-islands to infer likely promoters and we will add this information for comparison purposes. All of the annotations described here are with respect to a reference sequence, which, for the moment, is taken to be human. In future versions of our database we will also provide such annotations with reference to other vertebrate species. Given the huge number of possible promoter annotations, it will be important for the user to tailor the search and visualisation to specific elements of interest (e.g. particular transcription factor binding sites). In future implementations we will provide the possibility to filter all annotations with respect to additional contextual information.
Data Analysis
During pilot studies on human chromosome 21 it became clear that useful promoter predictions should also provide meaningful suggestions for primer selection. To overcome previous problems with genome-based primer design, we plan to map all BAC clones onto the genome and use computational predictions to suggest primers for a given promoter of interest. These primer predictions will also be made available to the NGFN partners through a web-based database interface.
Lit.: 1. Dieterich C et al. CORG: a database fro Comparative Regulatroy Genomics. Nuclear Acid Research. 2003; 31(1):55-7.