

Introduction
Nearly all hemato-malignant tumors are based on spe¬cific genetic aberrations, mainly reciprocal chromosomal trans¬lo¬ca¬tions. Chromosomal translocations are the reciprocal ex¬change of chromatide portions between two non-homologous chro¬mo¬somes. In general, this illegitimate recombination events are initiated at chromosomal sites where the DNA is damaged by radiation, apoptotic processes or cytotoxic sub¬stances (or metabolites thereof). By trying to repair these DNA lesions, the Non-Homolo¬gous-End-Joining (NHEJ) DNA repair pathway 1 is forming chromosomal translocations in a certain percentage 2. If genes are located at the sites of DNA damage, reciprocal fusion genes are generated. If the expression of these fusion genes give the affected cell any selective ad¬vantage, a pre-malignant clone is settled which can accumulate further mutations during rapid growth. Positive selection processes are then leading to the deve¬
¬The large population of leukemic cells are then causing fever, weakness, bone pain, platelet and red blood cell de¬pletion and hepato/splenomegaly.
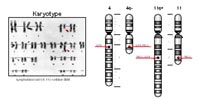
Our project is focussing on chromosomal translocations of the human MLL gene (11q23). The human MLL gene is frequently involved in chromo¬somal translocations and more than 50 different chromo¬somal translocations have so far been identified and characterized on the molecular level 3. All these MLL translocations are associated with acute (lympho¬blastic or myeloid) leukemia. The most frequent genetic aberration of the human MLL gene is the chromosomal translocation t(4;11)(q21;q23). These t(4;11) translocations are frequently found in infant (>75%), childhood acute leukemia patients (about 8-10%) and less frequent in adult leukemia patients (<1%). Due to the chromosomal trans¬lo¬cation, the MLL gene is reciprocally recombined with the AF4 gene, and thus, creating the MLL•AF4 and the AF4•MLL fusion genes (see Fig. 1). Both fusion genes are expressed into large proteins, but their functions are barely understood.
The wildtype MLL protein is part of an epigenetic machinery that is responsible for the activation of chromatin at specific target sites. For this purpose, the MLL protein is proteolyzed into the N-terminal MLL•N (p300) and the C-terminal MLL•C protein fragment (p180) that bind to each other to build the molecular platform for the association with several proteins 4. The MLL core enzymatic machinery is located at the very C-terminal portion, within the so-called SET-domain. The SET domain, in conjunction with ASH2L, WDR5 and RbPB5, allow histone H3 lysine 4 tri-methylation. Thus, histone core particles are modified in a mitotically stable fashion that result in the activation of chromatin. Recently, the MLL•C fragment has been shown to directly interact with MOF, a histone H4 lysine 16 acetyl¬transferase 5. This modification is important for certain aspects of active transcription pro¬cesses. The N-terminal portion is associated with several proteins, that in part provide specificity, as those proteins are able to bind in a sequence-specific manner to nuclear DNA (for review see 6). Therefore, any disruption of the amino acid sequence of the MLL protein and its reciprocal fusion to the large variety of different fusion partners is interfering with the biological function of the MLL protein. The presence of MLL fusion proteins seems to follow a dominant disease mechanism, as in the majority of investigated leukemia cells that carry MLL trans¬locations co-express the wildtype MLL protein and the reciprocal MLL fusion proteins.
Results/Project Status
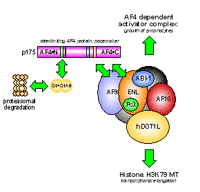
We study the biological functions of the MLL fusion partner AF4. The AF4 protein - when overexpressed in eukaryotic cells - is a very potent oncoprotein that lead to growth transformation and focus formation 7. Normally, the steady-state abundance of the AF4 protein is tightly regulated by a specific interaction with the E3-ubiquitin ligases SIAH1 and SIAH2. Both E3-ubiquitin ligases are able to bind specifically to a highly conserved protein domain located in the N-terminal portion of the AF4 protein (and the three other proteins of the AF4 protein family: LAF4, FMR2 and AF5, respectively). Due to the specific interaction with either SIAH protein, the AF4 protein be¬comes poly-ubiquitinylated and subsequently degraded via the proteasomal pathway. Therefore, very low amounts of AF4 are present in a normal eukaryotic cell that are below the detection level in Western blot experiments.
Transient or stable transfection with an AF4 expression con¬structs is sufficient to growth-transform cells. This classifies the AF4 protein as a novel and potent oncoprotein. The fusion of the AF4 N-terminal portion (about 400 amino acids) to the C-terminal portion of MLL (about 2600 amino acids) also create a potent oncoprotein, that accumulates in trans¬fected cells and efficiently leads to growth trans¬formation, comparable to the well known H-ras onco¬protein. From these data we concluded, that AF4•MLL fusion protein plays an important role for the pathological disease mechanism of t(4;11) leukemia 7.
By contrast, the MLL•AF4 fusion protein seems to be involved in cell cycle progression and regulation of apop¬tosis 8,9. Increasing amounts of the MLL•AF4 protein in the context of a normal cells leads to growth arrest and block of the apoptotic process. Experiments using the t(4;11) cell line SEM 10 and siRNA-mediated knock-down of the MLL•AF4 fusion protein is leading to enhanced apoptosis, loss of clonogenicity in semi-solid agar and survival of SCID mice after transplantation of siRNA-treated SEM cells. Unpub¬lished data from our own group demonstrated that the presence of both fusion proteins in a genetic background of a normal cell is strongly influencing cell cycle and a block og the apoptotic pathway 11.
Understanding AF4 protein functions
The human AF4 protein belong to a familiy of highly similar proteins, consisting of AF4, LAF4, FMR-2 and AF5. AF4, LAF4 and AF5 are translocation partner genes of the MLL gene and associated with the disease phenotype of acute leukemia. Transcriptional kncok-down of the FMR2 genby a triplet expansion mechanism is associated with a mild dorm of the Fragile X syndrome (FRAX E). Thus, all members of the AF4 protein familiy are involved in human diseases.
The members of this protein family are sharing highly homologous amino acid domains, called NHD, ALF, pSer, NLS and CHD 12. All members of the AF4 protein family are binding to both SIAH proteins. A specific 20 amino acid long protein sequence of the ALF domain is necessary and sufficient for binding. In addition, the NHD domain is providing a coiled-coiled domain that enhances binding to both SIAH proteins. Moreover, the AF4 protein - and presu¬m¬ably all other members of this protein family - makes part of a high molecular weight nuclear complex (AF4 multiprotein complex) which is composed at least by the AF9, ENL, AF10, ABI1, Pc3 and DOT1L proteins 13-15. (see Fig. 3). This complex links AF4 to trans¬crip¬¬tional processes, as the DOT1L protein is methy¬lating a specific lysine residue of the histone H3 protein 14. This modification is enhancing and maintaining transcriptional elongation processes of RNA Polymerase II, and thus, influences transcriptional process.
As the AF4 protein - and all other members of this protein family - is continously degraded, it becomes the rate-limiting component of this complex. Moreover, the AF4 protein is presumably a molecular trigger for cell growth as AF4 k.o mice show low amounts of lymphocytic precursors and growth retardation 16. By contrast, the abun¬dance of the MLL•AF4 fusion protein is much higher and enables the MLL•AF4 fusion protein to compete with AF4 for binding to the AF4 multiprotein complex.
Identification of AF4 target genes
By using these molecular informations, the following picture is emerging: AF4 or the AF4-multiprotein complex are both influencing transcriptional processes. In order to understand this function in more detail, we have begun to investigate this specific property of the AF4 protein. For this purpose, an AF4 k.o. and an AF4 wildtype cell line are used to extract mRNA. This mRNA fractions will then be used for Affymetrix Chip experiments to compare whole-genome-transcription profiles of both cell lines. So far, we have re-established the AF4 k.o. line in our laboratory, as this cell line was not further avail¬able from the published AF4 k.o. strain. The slow-growing AF4 k.o. cell line is currently being cultured and investigated by different experimental approaches. Within the next months, we should get a clear-cut picture about the potential targets of the AF4 protein by using isolated mRNA from wildtype and k.o. cells and the Affymetrix platform technology of the NGFN II network. These initial data will then be re-evaluated by using Q-PCR experiments and to identify interesting candidate target genes that can be further investigated.
Outlook
Having established the AF4 transcriptome, we plan further experiments by using an inducible MLL•AF4 transgene stably transfected into AF4 wildtype and knock-out cells. By comparing the expression profiles of these genetically modi¬fied cell lines, we hope to learn more about the oncogenic functions of the MLL•AF4 fusion protein, how it interferes with the wildtype AF4 protein and how trans¬criptomes are changed in the presence of particular MLL fusion protein. These data will enable us to understand this particular hemato-malignancy in more detail. This work will be accom¬panied by purification of the AF4 multiprotein complex and the two fusion protein complexes, AF4•MLL and MLL•AF4. From these data, we expect to gain sufficient insight into a pathological mechanism(s) that is able to con¬vert a normal hematopoietic precursor cell into a highly malignant leukemic cell. This knowledge could then be translated into ap¬proaches that addresses the pathological disease mecha¬nism mediated by the two reciprocal fusion proteins in order to develop novel therapeutic strategies.
Lit.: 1. Reichel M et al. Fine structure of translocation breakpoints in leukemic blasts with chromosomal translocation t(4;11): the DNA damage-repair model of translocation. Oncogene. 1998;17:3035-44. 2. Richardson C, Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697-700. 3 Meyer C et al. Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc Natl Acad Sci USA. 2005;102:449-54. 4. Hsieh JJ et al. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003;115:293-303. 5. Dou et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005; 121:873-85. 6. Popovic R, Zeleznik-Le NJ. MLL: How complex does it get? J Cell Biochem. 2005;95:234-42. 7. Bursen A et al. Interaction of AF4 wildtype and AF4•MLL fusion protein with SIAH proteins: indication for t(4;11) pathobiology? Oncogene. 2004;23:6237-49. 8. Caslini C et al. Modulation of cell cycle by graded expression of MLL-AF4 fusion oncoprotein. Leukemia. 2004;18:1064-71. 9. Thomas M et al. Depletion of the leukemic fusion gene MLL-AF4 by RNAi inhibits proliferation and induces apoptosis in t(4;11)-positive SEM cells. Blood. 2005. . [Epub ahead of print]. 10. Greil J et al. The acute lymphoblastic leukemia cell line SEM with t(4;11) chromosomal rearrangement is biphenotypic and responsive to interleukin-7. Brit. J. Haematol.1994;86:275-83. 11. Gaussmann et al., unpublished data. 12. Nilson I et al. The exon/intron structure of the human AF-4 gene, a member of the AF-4/LAF-4/FMR-2 gene family coding for a nuclear protein with structural alterations in acute leukemia. Brit. J. Haematol. 1997;98:157-69. 13. Srinivasan RS, Nesbit JB, Marrero L, Erfurth F, LaRussa VF, Hemenway CS. The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia. 2004;18:1364-72. 14. Okada Y et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167-78. 15. Zeisig DT et al. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene. 2005. [Epub ahead of print]. 16. Isnard P et al. Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood. 2000;96:705-10.