

Introduction
Oncogenic tyrosine kinases proved to be critical for survival and proliferation of malignant cells and were identified as therapeutic targets in a variety of neoplastic disorders. The availability of specifically acting small molecule kinase inhibitors instituted the era of targeted cancer treatment. In chronic myeloid leukaemia (CML), the impressive clinical activity of imatinib fundamentally changed treatment strategies. However, the development of clinical resistance toward imatinib constituted a major drawback in the treatment of advanced phase CML and Ph+ ALL.1 Mechanisms leading to resistance include amplification of the Bcr-Abl gene, acquired additional genomic alterations or, most importantly, specific mutations within the Bcr-Abl kinase domain that obviate binding to the drug target.2 Specific kinase domain mutations that cause resistance toward therapeutically applicated kinase inhibitors were also detected in hypereosinophilic disorders, gastrointestinal stromal tumors (GIST), in acute myelogenous leukaemia (AML), and in lung cancer. Novel kinase inhibitors display distinct profiles of resistance mutations within the same kinase, since different classes of compounds exhibit distinct modes of binding to the target kinase domain. Consequently, alternative Abl kinase inhibitors are capable of suppressing some Bcr-Abl mutations that cause imatinib resistance, as demonstrated for PD166326, BMS-354825, and AMN107. AMN107 and BMS-354825 recently entered clinical trials. Since data from analyses of resistant patients will be available not before a large number of patients will be treated, prediction of mutation patterns for compounds that are in preclinical or clinical evaluation is of utmost importance and represents a crucial step toward the development of tailored treatment strategies using sequential or combinatorial application of different kinase inhibitors.
Results/Project Status
A cell-based screening system allows identification of resistance mutations
We recently developed a cell-based screening strategy that employs the murine pre-B cell line Ba/F3, which was transformed by a specific oncogenic tyrosine kinase and then was cultured in the presence of a kinase inhibitor acting on that specific kinase.3,4 Colonies that grow under these conditions were expanded and analysed for mechanisms that determine resistance, such as kinase domain mutations of the Bcr-Abl gene. For this purpose, Ba/F3 Mig EGFPp185 wild-type cells were cultured in 96-well plates at a density of 4 x 105 cells per well in the presence of imatinib, or the alternative Abl kinase inhibitors PD166326 or AMN107. Different inhibitor concentrations were used: 2µM and 4µM for imatinib; 25nM, 50nM and 100nM for PD166326; 100nM, 200nM, 400nM for AMN107. Single colonies were picked and expanded for analysis. Resulting resistant sublines were cultured in the presence of inhibitor at a concentration corresponding to that used in the screen.
Novel Abl kinase inhibitors may be superior to imatinib
Imatinib was tested at concentrations that correspond to maximally achieved plasma concentrations in treated patients. In the presence of imatinib, Bcr-Abl kinase domain mutations occured at positions that are known from imatinib-resistant patients, and at novel positions.3 When applied to PD166326 or AMN107, the frequency of resistant colonies was lower when compared to imatinib. AMN107 and PD166326 both produced a limited set of kinase domain mutations when compared to imatinib, each displaying a distinct mutation pattern with a limited number of affected positions (see Fig. 1). Rarely emerging colonies at higher concentrations of AMN107 @ 400nM or PD166326 @ 100nM exclusively contained one single Bcr-Abl kinase domain mutation, T315I. This was in contrast to imatinib, where mutations were still widely distributed over the kinase domain at 4µM, which corresponds to the maximal achieved concentration in treated patients. Mutations identified in the AMN107 screen were recreated using site-directed mutagenesis and expressed in Ba/F3 cells. In cell growth assays, dose-response curves indicated that all mutations that were identified in the screen indeed mediate resistance to AMN107. However, all mutations that were identified in the screen, including exchanges frequently detected in CML patients with imatinib resistance such as P-loop mutations, were effectively inhibited when the AMN107 concentration was increased. The only exception was T315I, which was not suppressed by PD166326 or AMN107 at concentrations of up to 500nM PD166326 or 4000nM AMN107. Thus, in this cell-based screening system, an increase of the concentration of both second-generation inhibitors prevented the selection of resistant colonies, with the exception of Bcr-Abl T315I. Our findings indicate that PD166326 and AMN107 might be superior to imatinib in terms of the development of resistance.
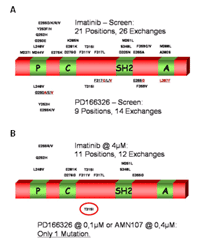
Analysis of sublines that did not contain Bcr-Abl kinase domain mutations
As observed in some imatinib-resistant CML patients, some resistant, non mutant sublines displayed increased Bcr-Abl protein levels, which are thought to increase the inhibitor concentration necessary for sufficient inhibition. Also, cytogenetic analysis of some resistant lines derived from the imatinib screen revealed novel cytogenetic aberrations, or amplification of the Bcr-Abl gene.4 Both findings were also reported in imatinib-resistant CML patients. Thus, this cell-based method is not only capable of producing clinically relevant point mutations that cause resistance of Ph+ leukaemia towards currently applicated and investigational drugs, but also bears the potential of revealing mechanisms of resistance other than Bcr-Abl point mutations. This strategy is therefore probably closer to in vivo conditions than a mutagenesis-based approach, which only allows selection of randomly introduced resistance mutations and, in addition can be performed with moderate efforts in time and labour.
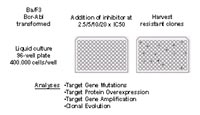
Outlook
Therapeutic use of kinase inhibitors is possible whenever growth and survival of a disease is dependent on the activity of a specific oncogenic kinase. Resistance may occur as a result of specific kinase domain mutations that obviate binding of therapeutically applicated kinase inhibitors, as observed in patients with CML or Ph+ ALL and imatinib resistance. Specific mutations mediating imatinib resistance have also been described in FIP1L1-PDGFRalpha in patients with hypereosinophilic disorders, and were detected in the cKit kinase domain in patients treated with imatinib for gastrointestinal stromal tumors (GIST). Recently, a mutation in the FLT3 kinase domain has been described in a single patient treated with PKC412 for acute myeloid leukaemia (AML). It can be foreseen that resistance mutations in the FLT3 receptor tyrosine kinase gene will be identified in additional patients with AML and activating FLT3 mutations treated with different FLT3 inhibitors that are currently under clinical investigation such as PKC412, su11238 or CEP-701. Resistance mutations also evolve in epidermal growth factor receptor (EGFR) in patients with non-small cell lung cancer, and activating EGFR mutations who respond to the EGFR-inhibitor gefitinib. Our cell-based screening strategy proved robust, can be conducted with maintainable efforts in an academic setting, and more importantly, produces results that proved to be clinically relevant. In contrast to screening for drug resistant clones after random mutagenesis, our screening method is not limited to detection of resistance mutations in the target oncogenic kinase, but also identifies other changes that were reported in patients with inhibitor resistance, including amplification of the target gene, overexpression of the target protein and acquisition of cytogenetic abnormalities (Fig. 2). Thus, if a cell-based approach is used, known resistance mechanisms can be studied in detail and novel mechanisms of resistance can be identified for different targets using investigational compounds and clinically applicated drugs. This or similar screening approaches will provide data that can be translated into combinatorial and sequential treatment strategies for a variety of rational drug targets in hematology and oncology. Specific patterns of resistance mutations can be predicted prior to their identification in patients, and critical serum concentrations can be defined that have to be achieved in the clinic in order to minimize the emergence of resistance. We are currently applying this method to other diseases that have been successfully treated with kinase inhibitors. Specifically, we are currently conducting resistance screens for lung cancer, acute myelogenous leukaemia and hypereosinophilic syndrome.
Lit.: 1. von Bubnoff N et al. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359:487-491. 2. von Bubnoff N et al. Resistance of Philadelphia-chromosome positive leukemia towards the kinase inhibitor imatinib (STI571, Glivec): a targeted oncoprotein strikes back. Leukemia. 2003;17:829-838. 3. von Bubnoff N et al. A cell-based screen for resistance of Bcr-Abl-positive leukemia identifies the mutation pattern for PD166326, an alternative Abl kinase inhibitor. Blood. 2005;105:1652-1659. 4. von Bubnoff N et al. A cell-based screening strategy that predicts mutations in oncogenic tyrosine kinases: implications for clinical resistance in targeted cancer treatment. Cell Cycle. 2005;4:400-406.