

Introduction
RNA interference (RNAi) is an evolutionary well conserved mechanism for post-transcriptional gene silencing. Once incorporated into a protein complex with nuclease activity (RNA induced silencing complex, RISC), short double stranded RNA molecules (Small interfering RNAs, siRNAs) can serve as a guide to recognize and cleave RNA molecules harbouring a homologous sequence. During the last years, RNAi has turned to be an indispensable tool not only for the functional analysis of cancer related genes but has also been successfully applied for the inhibition of tumour cell proliferation in vitro and in vivo. These findings suggest that, due to the specificity and efficacy of siRNA ,mediated gene silencing, such molecules may hold great therapeutic potential. However, high costs for chemically synthesized siRNA molecules and delivery problems still hamper the broad applicability of RNA interference technology.
Results
To facilitate the applicability of RNAi within the CancerNet, we have established new RNA interference related tools and technologies. These include a procedure for the reliable, cost effective semi automated synthesis of siRNA molecules (patent pending) as well as the construction of sets of short hairpin (shRNA) expression vectors and a human siRNA database.
Enzymatic synthesis of siRNA molecules
We have established a procedure for the cost effective semi-automated enzymatic synthesis of siRNA molecules in 96well scale. Such siRNA molecules transferred into human leukaemia and colon cancer cells exhibited equally good gene silencing activity and specificity as chemically synthesized siRNA molecules at significantly reduced costs. A proof of principle experiment with 69 siRNA molecules synthesized in parallel demonstrated the high, and more importantly, comparable yields, underlining the robustness of our method. This technology has already been used to synthesize a set of mutants of a potent siRNA sequence for a systematic study of the effects of position dependent point mutants on siRNA knock down efficiency.
Human siRNA Database
Despite the strong progress made in the siRNA field during the last two years, gene knock down experiments still require a labour and cost intensive identification of potent siRNA sequences and the development of protocols for efficient siRNA delivery into the cell lines of interest that are frequently refractory towards siRNA incorporation. This prompted us to establish the human siRNA database HuSiDa (1). It provides sequences of published functional siRNA molecules targeting human genes and important technical details of the corresponding gene silencing experiments, including the mode of siRNA generation, recipient cell lines, transfection reagents and procedures and direct links to published references (PubMed), thereby supporting the setup and actual procedure of specific RNAi experiments in human cells.
pMACS-KK-H1: Magnetic enrichment shRNA-expressing cells.
High transfection efficiencies are an absolute prerequisite for successful knock down experiments. Unfortunately many cellular systems are hard or almost impossible to transfect. In collaboration with Miletnyi, Bergisch-Gladbach, we have constructed vectors that combine expression units for cell surface markers and shRNAs (short hairpin RNAs). This strategy allows the selective enrichment of transfected shRNA-expressing cells and the subsequent analysis of gene knock down phenotypes
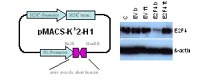
This example demonstrates the potential of the pMACS-KK2-H1 vector system. Magnetic enrichment of transfected cells is so efficient that an almost complete knock down of E2F4 can be observed in the enriched cell population (E2F4 b), although the transfected (cell surface marker and shRNA expressing) subpopulation represented only 5% of the total cell population. Therefore, this plasmid system enables gene knock down experiments in cells that are hard to transfect without demanding optimization of transfection conditions or purchasing expensive electroporation equipment.
This strategy can yield sufficient material for subsequent mRNA preparations for Affymetrix chip hybridization experiments and even for extensive biochemical analyses. The availability of pMACS-KK2-H1 vectors harboring positive and negative control inserts and detailed magnetic selection protocols as well as the user friendly designed shRNA insert cloning strategy that is compatible with the popular pSuper vectors and their derivatives are additional advantages of this plasmid based RNA interference system.
pRep-H1: long term gene silencing with replicating vectors
Silencing of essential genes will result in obvious phenotypes, easily detectable in standard culture conditions after transient transfection of siRNA molecules. In contrast, functional analysis of modulating genes (modifiers) requires more sophisticated assays monitoring growth factor dependence, responses to genotoxic stress etc.. Such assays are often incompatible with transient transfection of siRNA molecules that often requires high cell densities that can result in cell confluency 48 to 72 hours after transfection. Even worse, signal transduction pathways activated by transient transfection of siRNAs can act synergistically with genotoxic stress induced signal transduction pathways. Therefore, long term gene silencing with shRNA expressing vectors is the method of choice for the functional characterization of nonessential genes. Unfortunately, stable transfection of conventional shRNA expression vectors has severe disadvantages. In most cases, only a very small fraction of transfected cells (>0.1%) will integrate transfected plasmids into their genome. Even worse, not all antibiotic resistant cells will express shRNAs. Therefore, cell clones have to be picked and characterized. In addition to the extra effort, the usage of cell clones can also complicate the identification of target genes, because already randomly picket clones from untransfected “homogeneous” cell populations may display severe differences in gene expression patterns.
In an attempt to establish alternatives to retroviral vectors for the use in non S2 laboratories, we have constructed replicating hairpin expression vectors for long-term gene silencing in mammalian cells.
The performance of the pRep-H1 vector has been extensively tested in several cell lines, including K562, LN229, HEK293, and U373. In collaborations with several other CancerNet-members, it was successfully used to generate numerous pools of stably transfected cells for the functional analysis of cancer related genes.
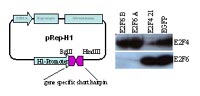
As shown in Fig. 2, the replicating, shRNA expression vector pREp-H1 mediates efficient long term knock down of target genes in mammalian cells (>4 weeks in continuous culture tested). pRep-H1 transfected cells are unable to separate antibiotic resistance and shRNA expression, because first, even after 4 weeks the complete pool of antibiotic cells still displays almost quantitative gene silencing and second, all our attempts to generate antibiotic resistant K562 cell pools expressing shRNA directed against BCR-ABL-kinase, essential for K562 cell survival, have failed (data not shown). This eliminates the need for picking and characterization of cell clones with all the aforementioned associated disadvantages. Transcripts of some shRNA expression vectors, especially such based on the U6 promoter, have been described to induce interferon response. This limits the applicability of such vectors. The comparative Affymetrix array analysis of the expression profiles of HEK293 cell pools stably transfected with pRep-H1 expression with or without shRNA-inserts using did show not any evidence for a shRNA specific activation of interferon response genes.
Consequently, pRep-H1 seems especially suited for long term gene silencing in mammalian cells. Its user friendly designed shRNA insert cloning strategy that is compatible with the pSuper vectors and their derivatives and the availability of vectors already containing positive and negative control shRNA-inserts make it an attractive alternative to existing retroviral shRNA expression vectors.
pFlp-H1: FLP-In shRNA expression vector
One disadvantage of replicating vectors is that their maintenance requires continuous selection pressure. Therefore, these vectors are not suited for the generation stably transfected cell pools to study gene function (metastasizing, response to genotoxic stress etc.) in xenograph mouse models. This prompted us to construct the pFlp-H1 vector. It enables the generation of isogenic cell pools by Flp-recombinase mediated, site specific integration of a shRNA expression cassette thatara also especially qualified for Affymetrix array analysis of knock down experiments.
Services and collaborations
For NGFN2 members, the proprietary shRNA expression vectors are available on request, including all necessary positive and negative control constructs. As a special service for NGFN2 members, we also offer to generate pools of cell lines, stable transfected with our replicating vector pRep-H1 and its derivatives for the functional analysis of nonessential genes in cell culture. This includes assistance in the design of shRNA sequences, cloning of shRNA coding inserts, sequencing, transfection into the cell line provided by the collaborator, the generation of stable transfected cell pools and the monitoring cell proliferation during the initial antibiotic selection period to detect and to document proliferation associated knock down phenotypes followed by shipping the cell pools to our collaboration partners for detailed characterization.
Outlook
We will continue to support CancerNet RNA interference users by providing informations about functional siRNA sequences, suitable recipient cell lines and validated transfection protocols. We will also continue to provide our “siRNA transfection efficiency control kit” that includes positive and negative siRNAs (both, synthetic siRNAsand shRNA expression vectors), real time PCR primers and detailed protocols for siRNA knock down experiments.
We are trying to advance siRNA delivery methodology focusing on alternatives to conventional siRNA transfection reagents.
We are also continuously improving our shRNA expression vectors. Derivatives of pRep-H1 with alternative antibiotic resistance genes and/or co-expressing red fluorescent protein have already been constructed and are under examination. The main focus here is on the development of Cre-lox and tetracycline regulated shRNA expression vectors that will enable the functional characterization of any gene in pools of stable transfected cell populations.
Lit.: 1. Truss et al., HuSiDa—the human siRNA database: an open access database for published functional siRNA sequences and technical details of efficient transfer into recipient cells. Nucleic Acids Res. 2005 Jan 1; 33 Database Issue:D108-11. 2. Wiebusch et al., Inhibition of human cytomegalovirus replication by small interfering RNAs. J. Gen. Virol. 2004 Jan; 85 (Pt 1): 179-84. 3. Tchernitza, O. et al., Transcriptional basis of KRAS oncogene-mediated cellular transformation in ovarian epithelial cells. Oncogene 2004 June 3 ; 23 (26): 4536-55.