

Introduction
The transition to malignancy requires an extensive reconfiguration of the genome´s expression program that does not result entirely from direct changes in primary DNA sequences (mutations), but from chromatin remodeling, histone modification and DNA methylation. DNA methylation of promoter regions has already been described to be causally involved in the silencing of tumor suppressor genes in cancer. Current data support that histone H3 lysine 9 (H3K9) trimethylation that correlates with gene silencing can promote DNA methylation and may be the initiating event in gene silencing. The heterochromatin protein 1 (HP1) recognizes H3K9 trimethylation and recruits further histone methyltransferases and interacts with components of the DNA methylation machinery, which might trigger sequential events leading ultimately to transcriptional repression. In addition, methylation of clustered repetitive elements in large chromatin segments not only contributes to specialized structures in pericentric heterochromatin, but also seems to be essential for the regulation of genome stability. Mobilization of these regions facilitates recombination between nonhomologous loci, leading to chromosomal deletion and translocation. Recent evidence indicates that loss of histone 4 lysine 20 (H4K20) trimethylation at constitutive heterochromatin in certain cancer cells is associated with hypomethylation of DNA at these sequences and this might be important for genomic instability.
The goal of this project is to determine specific histone methylation marks in the whole genome characteristic for transformed cells by using chromatin immunoprecipitation (ChIP) and differential cloning. Antibodies recognizing specific histone marks are used to analyze the function of histone methylation in cell transformation in a systematic genome-wide analysis in this project. The global analysis of epigenetic modifications on the basis of histone methylation by ChIP will complement the currently done global gene expression profiling and genome-wide DNA methylation analysis by other groups in the NGFN2 cancer net. This approach can lead to the identification of master regulator genes of the global gene expression profile, which should have the highest relevance as therapeutic targets.
Results/Project Status
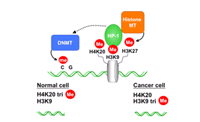
Characterization of histone marks in cancer cells
DNA methylation, chromatin structure, and gene silencing are interconnected in mammals. High levels of CpG methylation and hypoacetylated histones coincide with heterochromatin. In addition to these global levels specifc modifications on histone H3 namely trimethylation of lysine 9 and 27 and histone H4 lysine 20 mark silenced chromatin regions. These modifications are found in repetitive elements, noncoding regions as well as in promoter and coding regions of silenced genes. It will be important to identify differences in histone modification in cancer cells in all these regions, which might be causative in transformation.
We use antibodies specific against lysine trimethylation (H3K9, H3K27, H4K20) in ChIP to isolate genomic fragments which are silenced in tumor cells. Cancer cell lines from different tissue (breast, lymphoma, leukemia) are used in these experiments and repetitive regions as well as coding regions known to be silenced are analyzed by quantitative PCR in the precipitated samples. We further concentrated our work on cell lines derived from classical Hodgkin lymphoma (cHL). The basic transforming events of these cancer cells, which are derived from germinal centre B-cells, are still unknown. We observed that these cells are dependent on distinct components of the JAK/STAT signaling pathway, which is normally tightly regulated, but constitutively active in these tumor cells. Transcriptional regulation by STAT proteins is essential for cell survival and proliferation of cHL cells. Microarray analysis identified several genes, which are down regulated by the JAK/STAT pathway. However, transcriptional activation of negative regulators of the JAK/STAT pathway seems to be disturbed in some cHL cells (Baus and Pfitzner, 2005). The contribution on histone lysine methylation and DNA methylation in transcriptional repression and tumorigenesis is currently under investigation.
Cloning of genomic fragments containing histone methylation marks of silenced chromatin
In order to isolate novel sites of epigentic inactivation by histone methylation in cancer cells, we are using a modification of the ChIP method, which allows us to isolate all genomic fragments (promoters, enhancers, coding regions, noncoding regions) containing specifically modified histones. Chromatin fragments are therefore precipitated with antibodies specific against the lysine methylation marks of silenced chromatin (see above) from tumor cells, ligated to linker sequences, amplified by PCR and cloned into vectors to generate libraries of epigenetic modified genomic fragments. To identify genomic fragments, which are characteristic for tumor cells, differential hybridization against genomic fragments isolated by ChIP from non-transformed B cells are included in the cloning procedure. The libraries are currently generated and evaluated for inserts, which are characteristic for tumor cells.
Correlation of histone modification and DNA-methylation
It has been proposed that histone methlyation that correlates with silent chromatin also facilitates DNA methylation in these regions. To determine a correlation between histone modification and DNA methylation, fragments precipitated with antibodies against trimethlyated histones are ligated to specific linkers, amplified, fluorescently labeled and hybridized with CpG island arrays. This part of the project is currently done in cooperation with the SMP Epigenetic, PEG-S04T07 CpG-island microarrays, by Dr. Waha, University of Bonn. Positive CpG island clones will be sequenced and DNA-hypermethylation of these regions will be determined by genom sequencing of bisulfid-treated DNA from cancer cells.
Identification and isolation of genomic fragments with characteristic epigenomic modifications from tissue
In order to isolate genomic fragments with characteristic epigenetic modification also from solid tumors it was essential to adapt the ChIP method and subsequent cloning of genomic fragments for tissue samples. For this we used as a model system mammary gland tissue from mice. The mammary gland undergoes cyclic processes of differentiation, which correlates with the expression of milk protein genes (#-casein, WAP). Epithelial components of the mammary gland are thought to arise from stem cells that undergo both self-renewal and differentiation. Deregulation of the self-renewal in stem cells/progenitors might be a key event in mammary carcinogenesis.
Mammary gland tissue from mice at the differentiation states: virgin, pregnancy, lactation and involution was isolated. ChIPs were performed with antibodies recognizing histone modifications characteristic for active genes (acetylated histones, trimethylated H3K4), for competent chromatin (dimethylated H3K4), silent chromatin (trimethlyated H3K9, H3K27) as well as antibodies recognizing transcription factors known to regulate milk protein expression (STAT5a, STAT5b, RNA-Polymerase II). We detected binding of transcription factors STAT5, RNA polymerase II and trimethylation of H3K4 only during pregnancy and lactation, when milk proteins are expressed. Significant H3K4 trimethlyation was observed in the coding regions of the genes, but not in the far upstream 5´ region. Enhanced trimethylation of H3K27 was observed at stages when the milk proteins are not expressed (virgin and involution) and spread over the whole gene. No trimethylation of H3K9 could be detected. Our results so far indicate, that it is possible to isolate genomic fragments from tissue samples by precipitation with antibodies against specific histone modification. We are currently testing if it is possible to generate a library from genomic fragments precipitated from mammary gland tissue. Isolated fragments are also used to identify regulated chromatin regions containing CpG islands by Chip on chip hybridization in cooperation with the group of Dr. Waha in Bonn. The analysis of the identified fragments will greatly expand the knowledge about epigenetic regulation of mammary gland differentiation and can also provide important information for further studies on mammary carcinogenesis.
Cooperation in the NGFN2
We are cooperating with projects in the NGFN2 cancer net as well as SMP Epigenetic: With the project of Prof. Heinzel, university Jena predictive and pharmacodynamic markers for cancer therapy will be identified. ChIP with antibodies against methylation marks will be used to determine the effect of VPA on histone methylation of specific genes already identified to be derepressed by VPA. In cooperation with the project of Dr. Waha, university Bonn epigenetically down-regulated tumor suppressor genes will be identified in gliomas. Therefore glioma cell lines, which achieve chemoresistance will be analyzed by ChIP on CpG island Chip arrays. In a cooperation with Prof. Marschalek, university of Frankfurt we are analyzing histone modification at the breakpoints of the MLL gene, which is associated with high-risk acute leukemia.
Further cooperations are planed with Dr. Radlwimmer, SMP-DNA on the preparation of customized Chips representing genomic regions regulated by histone methylation and Dr. Schuster SMP Epigenomics on the identification of DNA-methylation marks in DNA fragments identified as targets for histone methlyation.
Outlook
The aim of this project is the identification of epigenetically regulated genomic fragments characteristic for cancer cells by combining the methods of ChIP and differential cloning. This approach can lead to the identification of the master regulators in the process of transformation, which will be the most relevant targets for drug therapy.
Genomic fragments isolated by these methods will be validated for cancer specific histone modification, DNA-methylation and gene repression. Validated fragments will be used to generate a customized Chip representing genomic fragments repressed by histone methylation. This customized Chip can be used to identify epigenetic modifications in tumors of other entities by ChIP followed by hybridization of the Chip (ChIP on Chip) and can be used in further extensive studies of chromatin-modification with other projects from the NGFN2.
Epigenetic changes identified in this project might in addition offer unique prospects for cancer diagnostics. ChIP has been an extremely useful research tool to analyze chromatin protein composition and modifications in our hands. As DNA methylation often appears late in the process of epigenetic silencing, the identification of cancer relevant histone modifications might provide a valuable diagnostic tool for early stages of transformation. The potential reversibility of epigenetic states offers the opportunities for novel cancer drugs that can reactivate epigenetically silenced tumor-suppressor genes. DNA methyltransferases and histone deacetylases are already targets for epigenetic therapy. A detailed analysis of the outcome of such a therapy is important and can be done with the ChIP on chip technique using the customized Chip generated in this project.
Our analysis will therefore be highly relevant for several projects of the NGFN2 analyzing epigenetic modifications and altered gene expression in cancer. It should also be possible to transform the method to analyze epigenetic modifications, which correlate with other diseases investigated in the NGFN2.
Lit.: 1. Baus D and Pfitzner E. Specific function of STAT3, SOCS1 and SOCS3 in the regulation of proliferation and survival of classical Hodgkin lymphoma cells. Int. J. Can. in press.