

Introduction
The formation of high-molecular weight complexes (HMWC) by fusion proteins arising from chromosomal translocations is a hallmark of many leukemias. Cellular transformation and maintenance of the transformed phenotype are closely linked to the ability of the fusion proteins to form HMWC (1, 2). In the translocation t(9;22) the BCR/ABL protein associates via an N-terminal coiled-coil structure to form homo-tetramers. Disruption of this oligomerization domain results in a reduction of BCR-ABL autophosphorylation, increased STI-571 sensitivity and decreased transforming potential (3-5). Similarly, the translocation t(8;21), one of the most frequent chromosomal anomaly in leukemia, involves the AML1 gene (acute myeloid leukemia 1) on chromosome 21 and the ETO gene on chromosome 8. The ubiquitous expressed AML1 gene product acts as a transcription factor and belongs to the key regulators of hematopoietic cell differentiation towards the myeloid lineage (6). In contrast, ETO (also called MTG8, CBF2T1) is a transcriptional repressor mostly expressed in heart and brain tissue. The nuclear protein ETO has multiple domains for protein-protein interactions. In particular, molecules of the transcriptional repressor machinery like N-CoR, SMRT, mSin3A and HDAC1-3 make multiple contacts with ETO and contribute to the transforming properties of AML1/ETO (Fig.1).
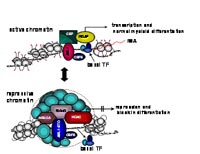
The nervy homology region (NHR) 2 within ETO is responsible for interaction with MTGR-1, ETO, ETO-2 and AML1/ETO itself. The NHR2 domain has a predicted coiled amphipathic helix structure and plays a crucial role for AML1/ETO induced oligomerization, HMW complex formation, repression of transcription and inhibition of myeloid cell differentiation. Apparently, the proper assembling of the transcriptional repressive machinery is one essential feature of HMW complexes as deletion of the oligomerization domain leads to almost complete loss of transformation potential. These and many other examples defined the oligomerization domains of oncoproteins as a central structural motif which is essential for transformation. The targeting of these structural domains by intercalating peptides could impair HMW complex formation and consequently could revert the malignant phenotype. Our work aims at the isolation of peptides from structural constrained peptide libraries targeted against the oligomerization domains of oncoproteins, in particular to the oligomerization domains of AML-ETO.
Results/Project Status
Recently we and others have shown that amino acids 335-432 of ETO, including the entire NHR2 domain, are essential for repressor activity and the ability to form high molecular weight complexes (7, 8). Based on these observations, we derived a peptide (NC128) aiming to interfere with the capacity of AML1/ETO to form HMW complexes. In our studies the NC128 peptide was found to bind specifically to the fusion protein AML1/ETO and to disrupt the formation of HMW complexes (Fig. 2).
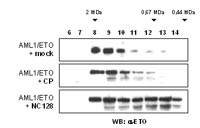
Binding of NC128 did not induce degradation of AML1/ETO protein nor interfered with the nuclear localization of the oncoprotein. Next we addressed the question, whether the peptide NC128 was able to release the transcriptional block induced by AML1/ETO. Total RNA extracted from the AML1/ETO transformed cell line Kasumi-1 was analyzed for expression of known AML1/ETO-target genes by RT-PCR. Transcription of the cell cycle regulatory genes p21 and p27 has been previously shown to be up regulated by AML1/ETO. Expression of NC128 in Kasumi-1 cells results in the down regulation of p21 and p27. This effect was specific for NC128, since expression of the control peptide did not alter p21 or p27 gene expression. In contrast, expression of the tumor suppressor p14ARF gene, which is specifically suppressed by AML1/ETO in AML blasts, was up-regulated in NC128 expressing cells suggesting that NC128 interferes with the transcriptional regulatory properties of AML1/ETO. Since Kasumi-1 cells share some common properties with hematopoietic progenitor cells including high expression levels of CD34 and c-kit and the ability to form colonies in soft agar, we investigated the effect of NC128 on the progenitor characteristics of Kasumi-1 cells. Typical features of progenitor cells, like high expression of the cell surface markers CD34 and c-kit, were lost in the presence of NC128. Furthermore, the clonogenic capability of Kasumi-1 cells was impaired in cells expressing NC128.
A further characteristic of AML1/ETO positive leukemia cells is the disability to proceed to terminal differentiation resulting in the accumulation of blasts and absence of mature granulocytes. Since this property of AML1/ETO correlates with its ability to form HMW complexes, we asked if expression of NC128 could influence differentiation of AML1/ETO transformed cells. In NC128 transduced Kasumi-1 cells we found a 20-fold increase in the number of cells expressing the myeloid differentiation markers CD11b and CD14, while no changes in differentiation pattern were observed in cells transduced with a control peptide.
Since recent studies revealed HDAC inhibitors as potent inducers of differentiation in leukemic cells we looked if NC128 could synergistically act with HDAC inhibitors in inducing differentiation of AML1/ETO transformed cells. Kasumi-1 cells expressing NC128 were treated with different concentrations of valproic acid (VPA). After 4 days treatment with VPA alone 15% to 30% of the Kasumi-1 cells expressed the myeloid differentiation marker CD13. However up to 70% of NC128 expressing Kasumi-1 cells did express CD13 after VPA treatment. Altogether these results demonstrate that expression of NC128 in combination with the HDAC-inhibitor VPA is effective in reverting the differentiation block induced by AML1/ETO. Finally, we could show that Kasimu-1 cells expressing the peptide NC128 are arrested in the cell cycle and undergo apoptosis. These effects were specific for NC128 and AMl1/ETO, since control cells negative for AML1/ETO expression were not affected by NC128 expression and control peptides did not influence the growth characteristics of Kasumi-1 cells (Fig. 3).
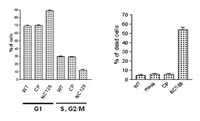
Our results imply that a functional AML1/ETO HMW complex is essential for the maintenance of the transformed phenotype and that disruption of AML1/ETO function, for example by intercalating peptides, results in cell differentiation and apoptosis.
Outlook
We propose the oligomerization domain of AML1/ETO as a valid target for a molecular intervention in t(8;21) leukemias. Within the translocation protein, the oligomerization domain plays a central role for the oncogenic properties of the fusion protein. As targeted disruption of AML1/ETO oligomerization leads to restoration of differentiation, growth inhibition and increased cell death in t(8;21) leukemic cells, small molecule compounds, developed to selectively block AML1/ETO induced high molecular weight complex formation, could provide promising tools to improve the therapeutic outcome of acute myeloid leukemias carrying the t(8;21) translocation. Toward this aim, we will screen libraries of small molecule compounds in order to find adequate inhibitors of AML1/ETO HMW formation. Similarly, retrovirally encoded peptide libraries will be screened for interference with AML1/ETO oligomerization. These studies will open this technology for multiple other applications.
Lit: 1. Minucci et al. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol Cell., 2000, 5:811-820. 2. Zhang et al. Oligomerization of ETO is obligatory for corepressor interaction. Mol Cell Biol. 2001, 21:156-163. 3. Zhao et al. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 2002, 9:117-120. 4. He et al. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood. 2002, 99:2957-2968. 5. Beissert et al. Targeting of the N-terminal coiled coil oligomerization interface of BCR interferes with the transformation potential of BCR/ABL and increases sensitivity to STI571. Blood. 2003 102:2985-2993.6. Kurokawa et al. Role of AML1/Runx1 in the pathogenesis of hematological malignancies. Cancer Sci. 2003;94:841-846. 7. Hildebrand et al. Multiple regions of ETO cooperate in transcriptional repression. J Biol Chem. 2001, 276:9889-9895. 8. Amann et al. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470-6483.