


Michal Janitz - Max Planck Institute for Molecular Genetics, Berlin - janitz@molgen.mpg.de
Introduction
The availability of the complete human genome sequence, with the advent of bioinformatics tools and array technologies, provide an excellent opportunity to unravel the identity and function of complex genetic networks and the underlying regulatory processes, which operate in the context of specific physiological responses, developmental switch points, and cellular events. Since the mid 1990s many research groups have focused on the analysis of individual promoters of genes to determine the function of even individual bases within the promoter elements for the transcription apparatus. But only since the availability of the complete human draft sequence (The International Human Genome Mapping Consortium, 2001) and the advent of large-scale expression profiling data on the genome-wide basis, has made possible the ability to correlate the transcript expression levels with the promoter elements of the respective genes (1,2). This research has just been started, and it will be essential to provide cloned material for as many promoters as possible to allow functional studies on these elements.
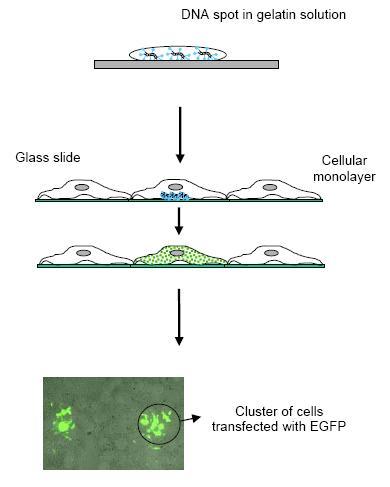 |
| Fig 1: Cell array technology combines the microarray technique with living cells. Reverse transfection allows for multiple, spatially separated transfections of nucleic acids in a single experiment. |
Since a simple map of promoter elements obtained by computer based analysis is not sufficient to understand promoter functions there is a need to functionally validate the obtained regulatory elements. At the Department of Prof. Lehrach we have established a cell array based high-throughput mammalian expression system (3,4) that allows the parallel functional analysis of thousands of promoter constructs. In this assay, the generated promoter-reporter vectors are printed at a high density on a glass slide along with a lipid transfection reagent. When the microarrays are covered with a layer of adherent cells only the cells growing on top of the spots become transfected, resulting in the expression of specific proteins in spatially distinctive groups of cells (Fig.1). The generated promoter-reporter constructs/library are tested in different cell types and under different environmental conditions (extracellular stimuli). Data collection is performed by digital image analysis at a single cell level and promoter expression levels is normalized relative to a cotransfected reporter vector (expressing high constitutive levels of Red fluorescent protein) (Fig. 2 and 3).
In first instance we plan to test the activity of the cloned promoters by performing reverse transfection into human or primate cell lines such as Hela, HEK293 and COS for which the transfection conditions have been optimized. For most of these cell lines we obtain a transfection efficiency of 10-20% when using cell arrays. By using similar standard cell lines derived from different tissues and by applying different extracellular activation protocols we hope to validate the predicted promoter activity of most of the clones. In a second phase we will analyze the tissue specificity of the promoters. Promoter activity is highly dependent on the cellular context in which it is introduced and transformed. Long term cultured cell lines often do not reflect the tissue anymore they were initially derived from. Therefore there is a need to extend this analysis to primary cell types. Since only small numbers of human primary cells can be isolated out of a tissue and the in vitro expansion of these cells is rather limited cell array offers a great advantage as compared to functional assays performed in microwell plate format. Standard microarray slides containing 5000 spotted expression vectors require as little as 1-3 10E6 cells. Furthermore by printing micro-arrays on smaller surfaces we can further reduce the number of cells significantly without loosing transfection quality (1-3 10E5 for an array of 1000 spots). Using primary skin fibroblasts for example we can obtain transfection efficiencies of 1-4% in TCA. Other primary cell types that will be analyzed besides fibroblast and epithelial cells are smooth muscle cells, endothelial cells, neuronal cells and macrophages. Knowledge of the transcription factor binding sites present on promoter elements is not necessarily an indication that the corresponding transcription factor is involved in the activity of this particular promoter. For example a perfect binding site located outside the appropriate context will bind its cognate protein but may not elicit any biological function in transcription. On the other hand, many promoters contain relatively weak binding sites that may be unable to bind their cognate protein on their own but such sites have been shown to be functional in many cases. Furthermore effector molecules that are acting upstream of a given promoter are in most cases unknown
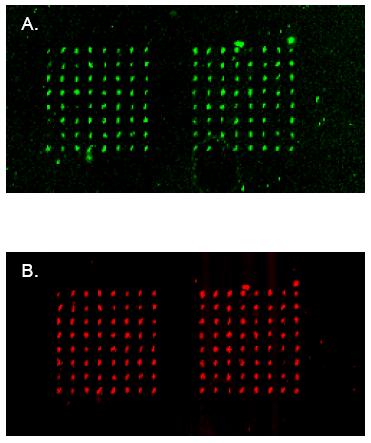 |
| Fig 2: An example of the signal detection for transfected cell array. Expression plasmid containing Green (A) or Red (B) Fluorescence Protein has been spotted in 8x8 spot block and subsequently transfected in an array format into the HEK cell line. Green or red fluorescence signal indicates clusters of cells expressing respective proteins. |
Results/Project Status
The goal of this subproject is evaluation and application of transfected cell array as a high throughput genomic technology platform for establishment of the cell type-specific patterns of mammalian promoter activities.
Promoter/reporter gene plasmid constructs will be spotted in an array format on the glass surface and subsequently transfected into the number of human cell lines as well as primary cells. The promoter activities will be measured using fluorescence detection of the reporter protein. Through the project-customized software the fluorescence signals from individual cells will be detected and normalized. It is envisaged to investigate 3000 human promoter regions. We have already successfully accomplished proof-of-principle studies where promoter activities have been measured on a cell array after normalization with the reference promoter activity (Fig. 3). At the moment the first set of 100 promoter fragments is evaluated in HEK and HeLa cell lines using this approach.
Only a small number of promoters is mapped experimentally. Today many theoretical as well as experimental approaches are going on to identify conserved regulatory regions. Currently more then 10,000 promoter regions have been identified. In NGFN-1 optimizing fund, the RZPD established a set of clones representing a minimal tiling path for the human genome, which will be used as templates for the promoter amplification. This set comprises 25,221 clones, nearly all of which are PAC and BAC clones that were used within the international effort to sequence to human genome. Therefore the insert sequence of the vast majority of the clones has been determined experimentally (22,982). In addition, clones were used, of which both ends were sequenced (BAC end scaffold map; 2239).
In order to identify which known or unknown molecules are responsible for the activity of validated promoter clones generated in the first part of the project we will cotransfect each promoter-reporter vector with an array of siRNA molecules specific for known or predicted transcription factors and signaling molecules (e.g. kinases, phosphatases). For this purpose we have developed the RNA Interference Transfection Array (RITA) so that either exogeneous (e.g. EGFP coding plasmids) or endogeneous (e.g. Lamin A/C) expressed genes can be specifically silenced (5). Concerning RNAi experiments the project will take an advantage from the resources of esiRNA against transcriptional factors created within SMP-RNA.
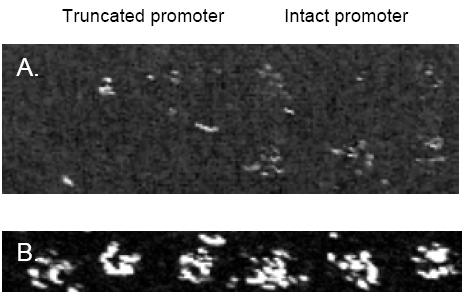 |
| Fig 3: An example of promoter activity measurement using reverse transfection. Deletion of the TATA box results in the decreased activity (left panel) of the heat shock protein 70 (HSP-70) promoter comparing to the intact promoter (right panel). Both promoters were cloned upstream of the EGFP gene. B: Expression of the red fluorescence protein used as a control reporter and cloned in the same vector as HSP-70 promoter. |
Outlook
Lit.: 1. Pugh BF et al.Genome-wide analysis of protein-DNA interactions in living cells. Genome Biol. 2001;2(4):REVIEWS1013. Epub 2001 Apr 4. 2. Iyer M et al. Two-step transcriptional amplification as a method for imaging reporter gene expression using weak promoters. Proc Natl Acad Sci U S A. 2001 Dec 4;98(25):14595-600. 3.
3. Vanhecke D, Janitz M. High-throughput gene silencing using cell arrays. Oncogene. 2004 Nov 1;23(51):8353-8. 4. Hu et al. High-throughput subcellular protein localization using cell arrays. Bichem Soc T. 2005, in press. 5. Vanhecke D, Janitz M. Functional genomics using high-throughput RNA interference. Drug Discov Today. 2005 Feb 1;10(3):205-12.